パーキンソン病のエンドトキシン仮説

国際パーキンソン病・運動障害学会
Movement DisordersEarly View
レビュー
オープンアクセス
パーキンソン病のエンドトキシン仮説
ガイ・C・ブラウン博士、マルタ・カマチョMS、キャロライン・H・ウィリアムズ=グレイMRCP、PhD
初出:2023年5月8日
https://doi.org/10.1002/mds.29432
関連する利益相反/金銭的開示: いずれの著者も、この論文に関連する財務上の開示や金銭的な利害の対立はない。
資金提供機関 G.C.B.のこの分野の研究は、英国医学研究評議会(MR/L010593)から資金を得た。C.H.W.G.は、医学研究評議会(MR/W029235/1)から資金提供を受け、Cambridge Centre for Parkinson-Plusから支援を受けている。また、NIHR Cambridge Biomedical Research Centre (NIHR203312)からも支援を受けている。表明された見解は著者のものであり、必ずしもNHS、NIHR、または保健省のものではありません。
について
セクション
要旨
パーキンソン病(PD)のエンドトキシン仮説は、リポポリサッカライド(LPS)エンドトキシンがこの疾患の病因に関与しているという考え方である。LPS内毒素は、グラム陰性菌の外膜に存在し、腸内などで放出される。PD初期の腸機能障害は、腸壁や血液中のLPS濃度を上昇させ、腸管神経細胞におけるαシヌクレインの凝集と末梢の炎症反応の両方を促進すると考えられています。血中のLPSやサイトカイン、腸-脳軸を介した脳への伝達は、神経炎症とα-シヌクレイン病態の拡大をもたらし、脳幹核の神経変性と黒質におけるドーパミン作動性ニューロンの損失を悪化させ、PDの臨床症状として顕在化させる。この仮説を支持するエビデンスは以下の通りです: (1)腸の機能障害、透過性、細菌の変化がPDの初期に起こる、(2)PD患者の割合でLPSの血清レベルが上昇する、(3)LPSがα-シヌクレインの発現、凝集、神経毒性を誘発する、(4)LPSが末梢単球の活性化を引き起こし炎症性サイトカインの産生につながる、(5)血中LPSが脳の炎症とミクログリアによる中脳ドーパミン作動性ニューロンの特定の喪失に関与している、など。この仮説が正しいとすれば、治療法としては以下のようなものが考えられます: (1)腸内細菌叢の変化、(2)腸管透過性の低下、(3)循環LPSレベルの低下、(4)LPSに対する免疫細胞やミクログリアの反応のブロック、などが考えられる。しかし、この仮説には多くの限界があり、特にLPSレベルを下げることでPDの発症、進行、重症度を下げることができるかどうか、さらなる検証が必要である。© 2023 The Authors. Movement Disordersは、International Parkinson and Movement Disorder Societyに代わってWiley Periodicals LLCから出版されている。
1 はじめに
パーキンソン病(PD)は、先進国の65歳以上の約2%が罹患する一般的な神経変性疾患です。1-3 中脳のドーパミン作動性ニューロンの進行性喪失を特徴とし、動作の遅さ、振戦、硬直、姿勢不安定などの運動障害、さらに幅広い非運動症状を伴います。2.3 PDの主要な神経病理学的特徴は、レビー小体やレビー神経突起と呼ばれる神経内タンパク質凝集体の存在であり、その主成分はSNCA遺伝子由来の線維性α-シヌクレインである2。α-シヌクレインの凝集体は、腸の腸管神経細胞内にも存在し、迷走神経を経由して脳に伝播することが提案されている4。PD患者の約5%~10%は、単一の遺伝子変異によって引き起こされ、GBAとLRRK2の変異が最も一般的な遺伝子危険因子である3、5。この総説では、エンドトキシンがそのような要因の一つであるという仮説について概説する。
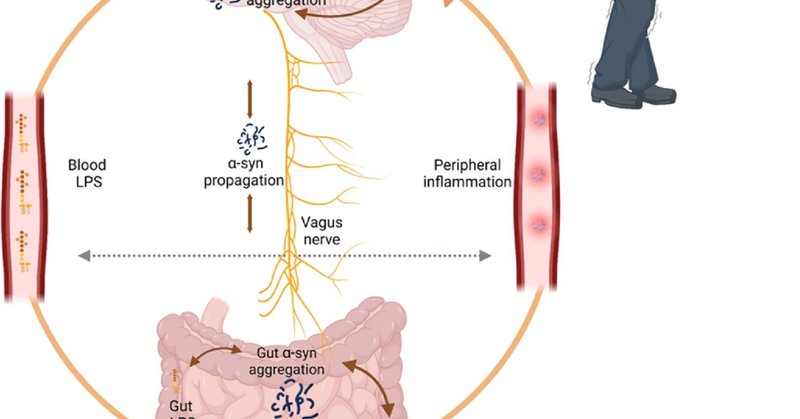
運動前期のPD患者の一部は、便秘、腸内細菌叢の変化、腸管透過性の亢進を患っている6, 7。このため、エンドトキシンが比較的良性の腸から循環血流に移行しやすく、エンドトキシンは炎症反応を引き起こし、脳にも影響を与える可能性がある。腸管由来のエンドトキシンは、α-シヌクレインの凝集を促進し、全身および脳の炎症を誘発し、脳のシヌクレイン障害と神経細胞の損失を悪化させる可能性があります6-8。当社および他の研究者による多くの既報は、エンドトキシンとPDとの関連を示していますが、ここでは、この理論の評価および検証を容易にするために、特定の仮説を、裏付け証拠およびその限界と共に概説します(図1)。
詳細は画像に続くキャプションに記載されています

図1
図ビューアーで開く
パワーポイント
キャプション
2 リポポリサッカライド内毒素とは何か、どこから来るのか?
リポポリサッカライド(LPS)は最も豊富な内毒素であり、内毒素という用語は一般的にLPSと互換性を持って使用されているので、ここではこれらの用語も互換性を持って使用することにする。LPSは、脂質A(通常は6本のアシル鎖が結合したリン酸化二糖)と、「コア」(修飾を受けた短い糖鎖)と、O-抗原(長さの異なる糖の長鎖)とが結合したものである(図2)10。LPSの脂質A成分はグラム陰性細菌の外膜の多くを構成し、O-抗原は細菌の表面を被覆する。LPSは生きた細菌から絶えず排出され、細菌が死ぬと放出される。10 LPSは細菌から放出されると、疎水性のアシル鎖のために小胞を形成するが、血液中のCD14やリポポリサッカライド結合タンパク質(LBP)に結合することにより、モノマーとして受容体に移行することができる11。
詳細は、画像に続くキャプションに記載

図2
図ビューアーで開く
パワーポイント
キャプション
人体内では、LPSの主な供給源は腸内細菌、皮膚、歯肉、肺などの上皮表面で、細菌感染によって増加することがある。6、7、12 健康な人におけるエンドトキシンの主要供給源はBacteroides fragilisや大腸菌などの腸内残留グラム陰性細菌である6、 7 腸壁を通過する量は限られており、そのほとんどは肝臓で除去されると考えられている。しかし、ほとんどのヒトの血液中には、少量だが有意なレベルのLPSが存在する。LPSは食品に含まれ13、高脂肪食の後にLPSの血液レベルが一過性に上昇(「代謝性内毒素症」)14、したがって食事は循環LPSレベルに影響を与える。また、肝疾患、肥満、腸管透過性のある患者でもLPS濃度は上昇する15-17。
腸は体内のLPSの主な供給源ですが、全身性の細菌感染によって血中LPS濃度が上昇し、PDリスクが高まることがあります。例えば、歯周炎(歯周病)は血中LPS濃度を上昇させ18、PDのリスクを1.4倍上昇させるが、感染を抑えるための歯のスケーリングもPDリスクを下げる。18-20 歯周炎の主要原因菌はPorphyromonas gingivalisで、血液や脳で見られる炎症性のLPSを産生する。18、21、22
3 LPSの病態生理
エンドトキシンは、特にTLR4(toll-like receptor 4)とその共受容体MD2を含む複数の受容体を介して、非常に低濃度で炎症を誘発することができる。11, 23, 24 CD14とLBPはLPSに結合し、TLR4への移行を助ける。 11, 23, 24 エンドトキシンによるTLR4の活性化は、NF-κB依存的に数百の炎症性遺伝子の転写活性化、インフラムソームの活性化、TNFα、IL-6、プロIL-1βといった炎症性サイトカインの放出を引き起こす25、 26 また、細胞内LPSは、ヒトではカスパーゼ-4またはカスパーゼ-5(マウスではマウスカスパーゼ-11)を直接活性化し、カスパーゼ-1を切断・活性化して、プロIL-1βをIL-1βに切断することがある27、28 場合によっては、カスパーゼ-1またはカスパーゼ-11により細胞膜を透過するガスデルミンDを切断・活性化し、パイロプシスにより細胞死を引き起こすこともある29、 30 エンドトキシンはまた、複数のスカベンジャー受容体に結合し、代替経路を介して補体を活性化し、補体受容体3を直接活性化し、神経毒性に寄与する可能性があります31-33。
10 LPSの脂質A成分は炎症を活性化するのに十分であるが、脂質A上のアシル鎖の数が6から5または4に減少すると、TLR4を活性化する能力が大幅に減少する34。実際、アシル鎖が4のLPS(Bacteroides doreiが産生するもの)は、MD2/TLR4を活性化せずに結合するため、アシル鎖が6のLPS(大腸菌が産生するもの)による活性化を抑制し、炎症を抑制できる34。したがって、腸内細菌によって、異なる形態のLPSが、炎症促進または抗炎症になる可能性はある。
自然免疫系は、エンドトキシンに対して迅速かつ強力な免疫反応を示します。なぜなら、エンドトキシンの存在は通常、細菌感染の兆候であり、感染が体内で広がるのを防ぐために、早期かつ強力な免疫反応が必要だからです。したがって、エンドトキシンに対する免疫反応は、通常、有益なものです。しかし、長年にわたるエンドトキシンに対する慢性的な免疫応答は、ここで提案するように、腸、体、脳の炎症性変化を引き起こし、PDを発症させる可能性があります。
4 PDにおけるエンドトキシンの供給源としての腸内細菌
腸管内腔にはLPSを含むグラム陰性菌が大量に存在し、血液中に放出されると致死的となる可能性がある7、12。早期PD患者の一部は腸管透過性が亢進しており、非公式に「リーキーガット」と呼ばれている。7, 35 リーキーガットは、エンドトキシンが腸壁に漏れることもあり、腸壁の炎症を引き起こすことがあり、エンドトキシン自体が腸管透過性と腸の炎症を誘発することがある36-38。腸の炎症は、動物モデルにおいて、腸の神経細胞におけるα-シヌクレインの発現と凝集を促進する(ただし、凝集はα-シヌクレインを過剰発現させた動物モデルで認められたことに注意)39。腸におけるα-シヌクレインの凝集は初期PD症例の大腸生検で観察されており、腸の感受性と運動性に影響を与えることを通じて、腸機能障害に寄与すると考えられます40。
便秘は運動症状に先立ち、PDの初期によく見られる特徴であり41, 42、少なくとも患者の一部では、腸の病理学的変化が疾患の初期症状の一つである可能性を裏付けています。PD患者の約50%が便秘であるのに対し、一般集団では約18%である。便秘はPDのオッズ比を2倍から10倍にする39, 43, 44。PDの早期便秘は、認知症発症の早さを予測する予後重要性を持つ45。
いくつかの研究で、運動症状と相関するPD患者の腸内細菌叢の変化が見つかっています39, 46-48 例えば、Goreckiら38は、非PD対照者の腸内細菌叢は74%がClostridia(良性のグラム陽性菌)、19%がBacteroidia(抗炎症性のLPSを産生)だったのに対し、PD患者のそれは33%がGammaproteobacteria、19%がVerrucomicrobiae(ともに炎症性のLPSを産生)だったと報告しています。Scheperjansら46は、PD患者においてLPSを産生するGammaproteobacteriaとEnterobacteriaceaeの割合が増加し、Prevotellaceaeが減少しており、これは腸管透過性の増加と相関している。Choiら49は、PDのいくつかのマウスモデルで腸内細菌科の細菌Proteus mirabilisが増加することを発見した。また、P. mirabilisを経口投与することで、腸と脳におけるα-シヌクレインの凝集、およびマウスにおける選択的なドーパミン作動性神経細胞の損失と運動障害を十分に誘発することがわかり、これはP. mirabilis由来のLPSに起因していることがわかった。Yanら50は、PD患者の腸内において、グラム陽性菌に対するグラム陰性菌の比率が大きく増加していることも発見した。PD患者では、運動症状の10年前までにグラム陰性ヘリコバクター・ピロリによる腸内感染と消化性潰瘍の発生率が高いことが報告されており51, 52、抗生物質によるピロリ菌の除菌はPD症状を改善した52, 53。
腸管内腔に生息する細菌種の変化は、上皮細胞同士をつなぐタイトジャンクションを制御することにより、バリア透過性に影響を与える36, 54, 55 したがって、腸内細菌叢の変化が、エンドトキシンが腸管透過性と炎症を高め、腸管ニューロンにおけるαシヌクレインの発現と凝集を誘発することによって、腸でPDを発症する可能性はある。しかし、腸内細菌叢の変化は、PDにおける他の腸の変化、例えば、腸の炎症、腸神経系の機能障害、腸の運動性の低下などの結果として生じる可能性もある。
5 PDでは血中エンドトキシン濃度が上昇する
血中エンドトキシン濃度は、表1に示す4つの研究において、PD患者および年齢をマッチさせた対照者において、異なる方法で推定されている。De Waalら56は、血小板の少ない血漿への抗LPS抗体の結合を定量化し、PD患者の平均LPSレベルがコントロールに対して約8倍上昇することを明らかにした。Loffredoら57は、抗LPS抗体を用いたサンドイッチ酵素結合免疫吸着法(ELISA)とLimulus amoebocyte lysate(LAL)アッセイの両方を用いて血清LPSレベルを定量化し、PD患者の平均LPSレベルの約2.4倍の上昇を確認しました。LAL試験は、カブトガニのリムルスの血球の凝固を誘導するLPS含有サンプルの生物学的活性を測定し、活性のエンドトキシン単位(EU)で定量化するもので、1EUはLPS100pgにほぼ相当しますが、これはLPS源によって異なります。私たちの研究8では、LALアッセイを用いて、PD患者の血清中の平均LPS濃度が約60%高いことを発見しました。Forsythら35もLALアッセイを用い、PD患者とコントロールの間で血清LPSに有意な変化を認めなかったが、彼らは特に便秘のPD患者を除外した。したがって、Forsythら35名の結果は、PDにおける血中エンドトキシン濃度の上昇は腸の機能障害と関連しているという説を支持するものである。
表1. パーキンソン病患者および対照者の血清または血漿中のリポポリサッカライド濃度を定量化した研究
研究内容 対照群におけるLPS PDにおけるLPS 方法
Forsyth et al (2011)35 0.82 ± 0.21 EU/mL 平均±SEM、N = 10 0.84 ± 0.13 EU/mL 平均±SEM、N = 9 血清中のLALアッセイ(便秘の患者は除外)。
De Waal et al (2018)56 0.51 ± 0.18 AU 平均±SD、N = 11 3.9 ± 0.7 AU 平均±SD、N = 11 抗LPS抗体の血小板貧困血漿への結合率
Loffredo et al (2020)57 12 ± 6 pg/mL 平均±SD, N = 64 29 ± 5 pg/mL 平均±SD, N = 8 血清中のLALアッセイおよびサンドイッチELISA
Wijeyekoon et al (2020)8 1.20 ± 0.64 EU/mL 平均±SD、N = 41 1.91 ± 1.66 EU/mL 平均±SD、N = 41 血清中のLALアッセイ法
注:平均値±SD/SEMは適宜表示。
略号 LPS、リポポリサッカライド、PD、パーキンソン病、EU、エンドトキシン単位、LAL、リムルスアメーバサイト溶解液、SEM、平均の標準誤差、N、サンプリング人数、AU、任意単位、SD、標準偏差、ELISA、酵素結合免疫吸着測定法(Erzyme-linked immunosorbent assay)。
これらの研究はいずれも比較的少数のPD患者(表1のN値)を用いたもので、最も大規模なものはPD患者41人と年齢をマッチさせた対照者41人を用いた我々の研究8である。この研究では、約25%のPD患者がコントロールのいずれよりも高いエンドトキシン値を示した。しかし、70%のPD患者の血清エンドトキシンは正常値であった(図3)。このことは、PDの生物学的異質性を強調するとともに、PD患者のサブグループにおいて、腸の機能障害と血清エンドトキシンの上昇が疾患発症に特に関連している可能性を示唆しています。これと同様に、PDにおける初期の消化器症状は普遍的なものではなく、新たにPDと診断された患者の約30%が便秘を訴えている58。今回の知見には検証が必要であり、エンドトキシン値が一過性に上昇するのか永続的に上昇するのか、また値が症状や疾患の進行と相関するのかどうかを明らかにするには、PD患者の血清エンドトキシン値の日、月、年単位での経時解析が興味深いと考えられる。
詳細は画像に続くキャプションに記載されています
図3
図ビューアーで開く
パワーポイント
キャプション
PDにおけるLPS濃度の上昇を示唆するこれらの興味深いデータにもかかわらず、ヒト血液中のLPS定量には多くの限界がある。まず、LPSの血中濃度は非常に低く、多くの市販のアッセイでは検出範囲以下である可能性があります。また、採血時(市販の採血管の多くはエンドトキシンを含んでいる可能性がある)、サンプル調製時、アッセイ試験時の汚染の可能性も大きな問題である59。Salden と Bas は、血液サンプルを直ちに 0℃に冷却し、サンプルを直ちに遠心分離することを推奨する59。血液凝固によりエンドトキシン活性がさらに失われるため、血清よりも血漿が好ましく、採血管は低濃度のヘパリンを含むことが望ましい59, 60 異なる測定法の技術的側面、測定法の感度比較、および異なる試料調製法については、他の場所で詳細に説明されている60, 61。
血液中のLPSを測定することは困難であり、血中LPSの半減期が短いため、全身エンドトキシンレベルの間接的なマーカーも使用されてきた。リポポリサッカライド結合蛋白(LBP)はエンドトキシンを中和し、血液から除去する働きがあるため、その値が低いほどエンドトキシンの曝露量または利用可能量が増加していることを示唆する62。複数の研究で、PD患者の血清または血漿LBPレベルが対照群と比較して低下していることが報告されており35、47、63-65、さらにレベルの低下はPDリスクの上昇に関連していることが判明している65。
6 末梢性エンドトキシンはPDに似た病態を誘発する
マウスやラットの腹膜にLPSを注射すると、慢性的な神経炎症、進行性の黒質病理、運動障害、黒質におけるドーパミン作動性ニューロンの特異的変性が引き起こされ、これは現在PDのマウスモデルとして使用されている。
72、73 この量(10μg)のエンドトキシンが全身に分布すると140pg/mLに相当し、PD患者の血清に見られる30pg/mLと比較すると、その差は大きい57。
健康なヒトにLPSを注射すると、痛み、疲労、睡眠増加、食欲不振、抑うつ症状など、多くの症状が(数時間)誘発される。しかし、これらの非運動症状の病態生理学的基盤は多因子であり、LPSによって誘発される病気行動は、他の多くの要因によって誘発される可能性があります。
7 エンドトキシンはマウスでα-シヌクレインと相乗効果を発揮できる
モデル系では、エンドトキシンはα-シヌクレインと相互作用または相乗作用して、さまざまな方法でPD病態を誘発することができる。例えば、マウスの腹膜にエンドトキシンを注射すると、大腸でのα-シヌクレインの発現とリン酸化が誘導され、腸の透過性が増加した37。エンドトキシンはまた、α-シヌクレインを、さらに自己持続的に線維化させる新しい形態に線維化させることができる76、77。対照マウスとA53Tα-シヌクレイントランスジェニックマウスにエンドトキシンを末梢注射すると、同様の急性神経炎が引き起こされたが、その後トランスジェニックマウスだけが持続的な神経炎、α-シヌクレインの凝集、レビー小体、黒質におけるドーパミン作動性ニューロンの進行性喪失を起こした79。同様に、ヒト A53T α-シヌクレインを発現させたマウスやLPSを単回投与したマウスでは、13ヶ月後に黒質におけるドーパミン作動性ニューロンの損失は見られなかったが、併用するとかなりの神経細胞損失が引き起こされた80。α-シヌクレイン過剰発現マウスでは、LPSの経口投与により運動症状が早期に発現した38。このことは、エンドトキシンの上昇と凝集性のα-シヌクレインがPDの神経細胞喪失を促進するという、PDの二兎を追う仮説を示唆している。
8 エンドトキシンは遺伝的なPDに関係する可能性がある
PDの約5%~10%は単発性、すなわち単一の遺伝子の変異によるもので、現在までにGBA、LRRK2、Parkin、DJ1、PINK1、SNCAなど少なくとも14の遺伝子が同定されている5。同様に、変異LRRK2は、末梢LPSによって誘発されるマウスの黒質におけるドーパミン作動性神経細胞喪失を増強する。81 GBA変異は、サイトカイン放出として測定されるLPSに対するマクロファージの炎症応答を増大させる82、 83 同様に、Parkinノックアウトマウスでは、LPS誘発の神経炎症、ドーパミン作動性ニューロンの損失、運動障害が増加し68、変異またはノックアウトDJ1では、LPS誘発のミクログリア活性化と培養およびin vivoでのドーパミン作動性ニューロンの損失が増加する。84、85 つまり、一般的に、PDに関連する遺伝子変異はLPSに対する炎症反応の増大と関連していて神経細胞の損失を促進している。したがって、PDのエンドトキシン仮説は、遺伝性PDと特発性PDの両方に関連する可能性がある。
9 エンドトキシンはPDの原因となる全身性炎症を誘発する
LPSは単球の強力な活性化因子であり、TLR4を介して結合し、下流のNF-κBおよびIRF転写因子の活性化、炎症性サイトカインおよびケモカインの産生を引き起こす。健康なヒトボランティアでは、LPSの静脈注射により血清TNFα、IL-6、IL-8、IL-10が誘導され、その後神経炎症が起こる。74、75 In vitro研究では、PD患者の単球はLPS刺激に対する反応が増加し、病気の重症度と相関することが示されている86が、これは普遍的な所見ではなく、病気のステージや性別によって異なる場合がある87。さらに、LPS受容体であるTLR4は、PDの血液由来単球や腸粘膜下層で発現量が増加しており、単球がLPSに反応するようにプライミングされている可能性を示唆している47。
炎症性サイトカインのレベルはPDの血清中で上昇し91、血中の免疫マーカーの炎症性プロファイルは、より速い疾患進行と関連している92。単球はこれらのサイトカインの主要な供給源であり、PD患者ではその表現型が変化し、貪食性と炎症性の高い古典的単球の割合が増加し、細胞の活性化と移動に関わる単球受容体の発現が増加している8、 86 これらの単球の変化は、認知症のリスクが高い人で最も顕著であり、自然免疫の活性化がより急速な疾患進行に寄与している可能性を示唆している。8 単球サブセットの変化は、「リスクのある」集団においてPD診断前に報告されており、自然免疫応答が疾患発症に寄与するという仮説を支持している93。
末梢のTNFα、IL-1β、LPSはそれぞれ、マウスの脳内でサイトカインやケモカインの放出を含む脳の炎症を誘発する。95 PDにおける末梢-中枢免疫クロストークのメカニズムはまだ完全に確立されていないが、エンドトキシン駆動の末梢炎症反応は、脈絡叢を介した免疫細胞の浸潤や血液脳関門(BBB)を通過するサイトカインの通過を介して脳の病理に影響を与えると考えられる96。
10 エンドトキシンは脳内に侵入し、ミクログリアを介した神経変性を誘発する可能性がある。
血液中のエンドトキシンがどのように脳に入るかは明らかではないが、血液中のエンドトキシンの濃度が高いと血液脳透過性が高まり、エンドトキシンやその他の炎症物質が脳に入る可能性がある97、98。99 血中エンドトキシンに反応した持続的な脳内炎症は、脳のTLR4を必要とし、血中サイトカインとは無関係であることから、エンドトキシンが脳に入り、そこでTLR4を活性化して神経炎症を持続させることが、少なくともマウスにおける急性LPS投与により示唆されている100。複数の研究により、PDの死後脳でTLR4の発現が増加していることが示されており、PDの脳はLPSに対してより敏感である可能性が示唆されています101-103。
同様に、LPSの慢性的な末梢注射により、マウスでは中脳ドーパミン作動性ニューロンがミクログリアを介し、かなり特異的に消失する33, 68-71
ミクログリアは脳のマクロファージで、脳における自然免疫と炎症の主要なメディエーターであり、PD患者の黒質ではミクログリアが活性化することが知られている。107-109 エンドトキシンはミクログリアの炎症活性化を直接誘導し、エンドトキシンによるミクログリアの活性化はマウスの黒質と同様に培養、110でドーパミン作動性ニューロンの死や消失を引き起こす33.68-71 例えば、マウスの腹膜にエンドトキシンを注射すると、脳のミクログリアが急性かつ永続的に活性化し、10ヶ月後に黒質のドーパミン作動性ニューロンが失われた。75 健康なヒトボランティアでは、1 ng LPS/kgを静脈注射すると、LPS注射3時間後に末梢ベンゾジアゼピン受容体(PBR)リガンドのポジトロンCT(PET)イメージングで測定した脳のほとんどの領域でミクログリアの強い活性化が起こった。
エンドトキシンによって活性化されたミクログリアは、複数の手段で神経変性を誘導する可能性がある。グリアとニューロンの共培養では、エンドトキシンはニューロンの損失を誘発し、ミクログリアの除去はこのニューロンの損失を防ぐ。110 IFNγの存在下で、LPSはグリアにおいて誘導性一酸化窒素合成酵素(iNOS)を誘発し、一酸化窒素は次にニューロンを殺すことができる111 特にこれが低酸素またはスーパーオキシドと結合した場合112.、 113 しかし、エンドトキシン単独では、神経細胞の壊死やアポトーシスは起こらず、むしろ生きた神経細胞をミクログリアが貪食し、貪食の結果、神経細胞が死滅する。115, 114 エンゲルハイムは、ストレスを受けた神経細胞からのウリジン二リン酸(UDP)放出を必要とすると考えられ、これがミクログリア上のP2Y6受容体を活性化してストレスを受けているが生存可能な神経細胞をミクログリアが貪食するきっかけとなった。例えば、P2Y6受容体ノックアウトマウスでは、末梢性エンドトキシンによるマウス黒質のドーパミン作動性ニューロンの減少が抑制された(図4)71。また、ラットの線条体にLPSを注射しても、神経細胞の減少が引き起こされるが、P2Y6受容体阻害剤によって抑制された115。末梢性エンドトキシンは、脳の古典補体系を活性化し、ミクログリアの貪食作用を活性化させ、神経細胞消失を引き起こすが、これは補体C3欠損マウスで防ぐことができる。33
詳細は画像に続くキャプションに記載されています
図4
図ビューアーで開く
パワーポイント
キャプション
エンドトキシン仮説に基づく11の可能な治療法
LPSがPDに寄与しているのであれば、以下に示すように、多くの治療戦略が臨床試験でさらに評価される可能性がある。しかし、LPSがPDのごく一部で上昇するのであれば、最も適切な患者を対象とするためには、患者の層別化が不可欠である。このような臨床試験において、血中LPS濃度をモニタリングすることは、臨床効果の測定と並んで、メカニズム原理の重要な検証を行うことになる。
(1) 腸内細菌叢を変化させる。炎症性エンドトキシン産生種を減少させるために腸内細菌プロファイルを操作することは、特定の抗生物質、経口細菌、または糞便微生物移植(FMT)を用いて達成することができる116。
(2) 腸管透過性を低下させる。118非ステロイド性抗炎症薬(NSAID)の使用は、逆説的に腸の炎症と関連している119。抗TNFα抗体は、クローン病患者の腸の炎症と透過性を減少させ、炎症が透過性増加の原因であることを示唆している121。メトホルミンはマウスで腸の透過性を減少させ123、PDに有益であると考えられる他の効果を持っている124。
(3) 血中のLPS濃度を下げる。これは、エンドトキシンに対するワクチン接種によって達成できるかもしれない。無毒化したLPSを動物に接種すると、抗LPS抗体が誘導され、その後のグラム陰性敗血症から動物を守ることができる125, 126 無毒化したLPSを牛に接種すると、初乳に抗LPS抗体が誘導され、ラットに与えることで血中LPS濃度を下げることができる127
(4) LPS受容体をブロックする。高血圧治療薬として認可されている既存薬であるカンデサルタンは、TLR4 の発現と活性を低下させることが示されており、BBB を通過することが期待され、安全性も良好であることから、パーキンソン病への再利用の候補となる。124、128 TAK242 は、TLR4 の強力な低分子阻害剤で、これまでに敗血症治療の臨床試験で使用し短期的に忍容性が高いことが示されている129。
(5) LPSに対するミクログリア反応をブロックする。TLR4の遮断に加え、エンドトキシンに対するミクログリア反応を抑制する戦略として、補体受容体3、P2Y6受容体、またはインフラマソームの遮断が考えられる33、71。
12 仮説とLPS測定法の限界
エンドトキシン仮説を支持する証拠を提供するために、齧歯類モデルが使用されてきた。しかし、マウスはヒトよりもエンドトキシンに対する感受性が低いため、ヒトが許容できるよりも高いレベルのエンドトキシンがマウスに注入されることになり、これらのモデルの生理学的関連性は不明である。
血中LPSの上昇もPDに特異的なものではなく、むしろ敗血症、肝疾患、歯周炎、筋萎縮性側索硬化症、アルツハイマー病など複数の病態で認められる。したがって、例えば、LPSとα-シヌクレインの発現・凝集、LPSとPD関連遺伝子の関連変異、LPSとインターフェロンの発現増加など、何らかの二重ヒット仮説が必要となる。これは、神経変性が脳を過剰なLPS反応に導くという考えに関連しており、内毒素血症のエピソードが、それ以外の独立した神経変性プロセスを悪化させる可能性がある132、133。
また、血中LPSの上昇がPDに必要でないことは、PD患者の一部(約30%)で血清LPSが上昇していること、便秘のないPD患者ではLPSレベルが上昇していないことから明らかである35。しかし、PDにおけるLPS測定は、単一のタイムポイントでの測定という横断的性質により制限を受けてきた。一方、マウス胚をLPSに胎内曝露すると、出生後に黒質のドーパミン作動性ニューロンが失われ、135、2ヶ月齢のマウスにLPSを単回腹腔内注射すると、10ヶ月後に黒質のドーパミン作動性ニューロンが失われる69。このことから、エピソード感染や腸機能障害によるLPS曝露により、中間時点でLPSレベルが上昇しなくても、いつかPDに至る可能性も考えられる。
血中LPS濃度の再現性と変化(同一患者で縦断的に測定)について、短期(数時間から数日)および長期(数年)で検証し、症状(例えば、腸の機能)や臨床的疾患の進行と関連付ける必要がある。また、PDの前駆期(例えば、急速眼球運動(REM)睡眠行動障害(RBD)を有するコホート)におけるLPSレベルを、腸機能およびPD転換リスクの評価と並行して測定することも有用であろう。
ヒト生体試料中のLPSレベルの測定は技術的に困難であり、LALアッセイやサンドイッチELISAを用いる市販のキットは、ヒト血液中のレベルに対して十分な感度を示さないことが多い136。さらに、いくつかの交絡因子が潜在している。例えば、血清中のアルブミンはLALを妨害する可能性がある137。血清の分離に必要な血液凝固もエンドトキシンの捕捉につながるため、血漿中のLPS定量が望ましい場合があり、血漿中のLPSレベルは血清中よりも高い138。食事は食事中のLPS産生細菌に影響し、血中エンドトキシンレベルは高脂肪食後に上昇する14。139 肝臓はエンドトキシンを除去する主要な部位であることから、肝臓の損傷もLPS測定に影響を与える可能性がある。13, 140 ワクチンはアジュバントとしてエンドトキシンを含むが、LAL測定に干渉する水酸化アルミニウムを含むことが多く、偽陰性になる:
エンドトキシンの定量は、理想的には血漿サンプルで実施されるべきである。
エンドトキシンの測定は、絶食の状態で行うべきである。
同時期の感染症を記録しておく
最新のワクチン接種から少なくとも1週間後に採血を行うべきである。
抗生物質の使用期間中は、採血を避けるべきである。
検体は直ちに0℃に冷却し、遠心分離を行う。
13 エンドトキシン仮説の結論と主要なテスト
PDのエンドトキシン仮説は、LPSレベルの上昇がPDの病因に関与していることを示唆している。腸管機能障害やリーキーガットにより、LPSが腸壁に入り込み、局所的なα-シヌクレインの発現や凝集を促進し、それが迷走神経を介して脳に拡散すると考えられる。腸管透過性の亢進は、血中のLPSの上昇にもつながり、末梢の自然免疫系や脳のミクログリアを活性化し、脳内のαシヌクレイン病態や黒質におけるドーパミン作動性ニューロンの喪失を促進すると考えられている。PDの臨床的・生物学的異質性を考慮すると、これらのエンドトキシン関連メカニズムは、この疾患の普遍的な特徴ではなく、患者のサブセットにおいて高度に関連している可能性があります。
エンドトキシン仮説には複数のバリエーションが考えられるが、これらすべての仮説の重要な検証は、エンドトキシンが上昇しているPD患者において、エンドトキシンを減らすことで疾患の進行速度を抑えることができるかどうかである。このような試験を行うには、ベースラインのエンドトキシンレベルに応じて患者を選択する必要があります。最初のステップとして、エンドトキシンレベルと疾患の発症および進行との関係を明らかにするために、大規模なPDコホートおよびプロドローマルコホートにおいてLPSおよびその関連マーカーを縦断的に定量する研究が必要である。
謝辞
オープンアクセスのため、著者らは本投稿から生じるすべてのAuthor Accepted ManuscriptバージョンにCreative Commons Attribution (CC BY) ライセンスを適用している。
著者の役割
すべての著者が原稿のデザイン、起草、編集に参加した。
オープンリサーチ
参考文献
PDFをダウンロードする
バックナンバー
国際パーキンソン病・運動障害学会ロゴマーク
リソース
ジュリー・ナッシュ
マネージングエディター
mdjedoffice@gmail.com
編集長
A. ジョン・ストエスル(CM、MD、FRCPC、FCAHS
ブリティッシュコロンビア大学
カナダ、ブリティッシュコロンビア州、バンクーバー
© 1998-2023 International Parkinson and Movement Disorder Society. すべての権利予約。
その他のリンク
ワイリーオンラインライブラリーについて
プライバシーポリシー
利用規約
Cookieについて
クッキーの管理
アクセシビリティ
ワイリーリサーチDE&Iステートメントとパブリッシングポリシー
ヘルプ&サポート
お問い合わせ
トレーニングおよびサポート
DMCAと海賊版の報告
オポチュニティ
サブスクリプションエージェント
広告主・企業パートナー
ワイリーとつながる
ワイリーネットワーク
ワイリープレスルーム
著作権 © 1999-2023 John Wiley & Sons, Inc. 無断転載禁止ワイリーホームページ
この記事が気に入ったらサポートをしてみませんか?