サリチル酸の合成にナトリウムフェノキシドを用いるのはなぜか。~イオン半径と位置選択性~

本稿の要約
ナトリウムフェノキシドと二酸化炭素を用いたKolbe-Schmitt反応によるサリチル酸の合成がオルト位選択的に進行するのは、ナトリウムイオンのイオン半径が丁度いい大きさだからである。この反応の進行はアルカリ金属イオンによる二酸化炭素分子の活性化がキーであるが、ナトリウムよりも下の周期のアルカリ金属フェノキシドではイオン半径の増加によりパラ位でのPh-CO2反応も活性化することができるようになるためパラ置換体の副生が多くなる。リチウムフェノキシドが用いられないのはおそらく単純な化合物の安定性と扱いやすさの為であろう。
初めに
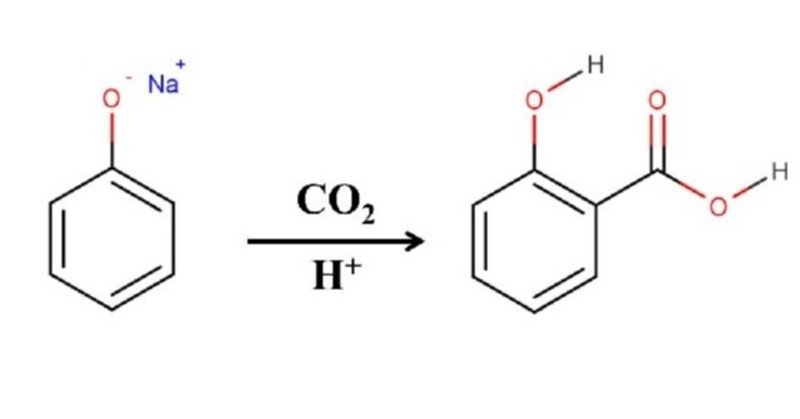
Kolbe-Schmitt反応をご存じでしょうか。反応名だけ聞いてもピンとくる方は多くないでしょうが、フェノールのアルカリ金属塩に二酸化炭素を加圧下反応させて芳香環にカルボキシ基を導入させる方法と聞けば思い出せるかもしれません。これはサリチル酸を合成する方法として高校有機化学でも習う重要反応です。反応式は図1のようになります。
図1. Kolbe-Schmitt反応によるサリチル酸の合成
この反応、高校化学の段階ではただの覚えなければならない反応の一つにすぎませんが、よくよく見てみると実に不思議な反応です。まずこの反応は有機反応にしては珍しく二酸化炭素が反応試薬となっています。無機化学では水酸化カルシウムないし炭酸カルシウムと二酸化炭素との反応を習いますが (石灰水に二酸化炭素を吹き込むと白濁するやつ)、高校までの有機化学で二酸化炭素を試薬として使うのはおそらくこの反応が唯一ではないでしょうか 。大学での有機化学でも二酸化炭素を試薬として用いる反応を習う機会はそう多くありません。 その点で特異な反応といえます。また、この反応はオルト1置換体が主生成物というのも不思議です。単純に考えればこの反応は反応点が3つ(イプソ位も考えれば4つですが今回は考えません)あり、1置換の場合はオルトメタパラの3種類が生成物となる可能性があります(ナトリウムフェノキシドは対称なので1,5位のオルトと2,4位のメタは同じ)。また、1置換だけでなく、例えばオルトとメタに1個づつ置換したりオルト2置換なんて生成物だってあってもよさそうです。そういうもんだといってしまえばそれまでですが、どういう理屈でこの反応は進行しているのでしょうか。改めて考えてみましょう。
単純な反応機構を考えてみる
Kolbe-Schmitt反応は単純に考えれば図2のような芳香族求電子置換反応と考えることができます。
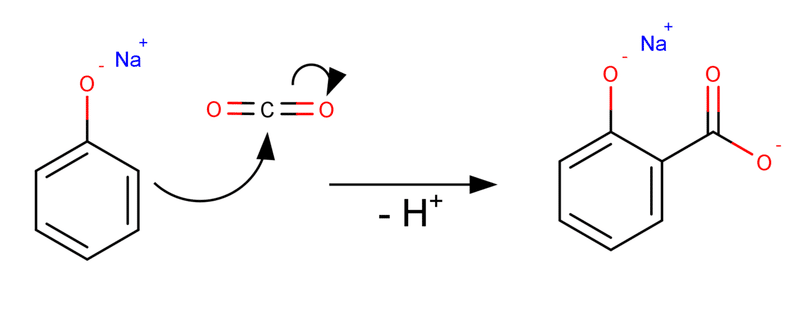
図2. 単純に予想されるKolbe-Schmitt反応の反応機構
図2における曲がった矢印は電子の動きを示しています。すなわち、δ+に分極している二酸化炭素の炭素が芳香環を求電子攻撃してカルボン酸が形成されるという流れです。この反応機構が正しいとすると、1置換体しか生成しない理由がすぐ分かります。芳香環にくっついたカルボニル基は電子吸引性不活性化基として振る舞います。すなわち反応の源である芳香環の電子をカルボニルが吸ってしまいますから、次の求電子置換反応が進行しづらくなります。これはFriedel-Crafts アシル化反応でアシル置換体が1つしか生成しない理由とほぼ同じといえます。
しかしながら、そう考えるとオルト位選択的な理由が微妙です。確かにナトリウムフェノキシドで芳香環に結合している酸素アニオン(フェノキシドイオン)は電子供与性のオルト‐パラ配向性活性化基ですから、オルト位で反応が進行するというのはわかります。が、ならパラ位でもある程度進行してしまうはずですし、何なら立体障害の少ないパラ位の方が優先したっておかしくはありません。そうなると、どうもこの反応は図2のような単純な話ではないように思えます。
アルカリ金属イオンの違いによる生成物の変化
Kolbe-Schmitt反応が図2のような単純な反応と考えることができない理由がもう一つあります。それはフェノキシドに対応するアルカリ金属イオンの違いによって生成物が異なってくるというものです。実はKolbe-Schmitt反応によるサリチル酸の合成においては、リチウムフェノキシドもしくはナトリウムフェノキシドを基質として用いた場合のみほぼオルト置換体が生成します。ナトリウムよりも下の周期のアルカリ金属イオンではパラ置換体の生成が無視できなくなり、生成物に占めるパラ置換体の割合は下の周期のアルカリ金属ほど多くなっていきます。
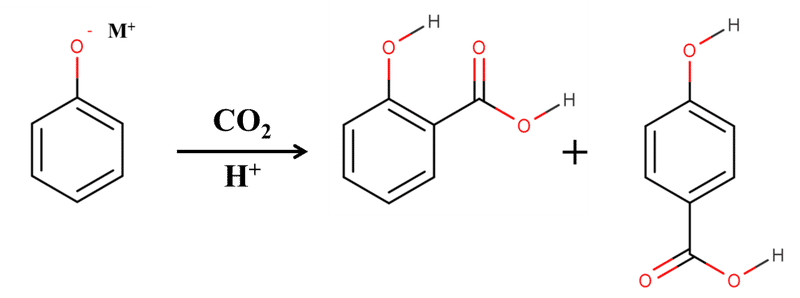
図3. M=K,Rb,Csの時のKolbe-Schmitt反応
すなわち、Kolbe-Schmitt反応によるサリチル酸の合成において、生成物に占めるパラ置換体 (p-ヒドロキシ安息香酸)の割合は各イオンごとに
Li ≦ Na < K < Rb < Cs
の順で増加していくのです。このことは、どうもこの反応の進行にはアルカリ金属イオンが深くかかわっていることを示しているように思えます。
アルカリ金属イオンが二酸化炭素を活性化する
実は当初、ナトリウムフェノキシドと二酸化炭素のKolbe-Schmitt反応は図4のようなO-カルボキシル化からのオルト位分子内転移によって進行すると考えられていました。
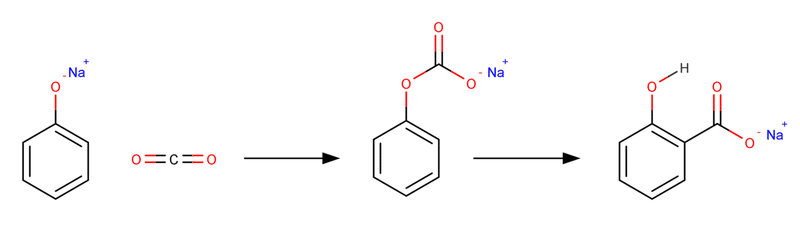
図4. 当初考えられていた反応機構
しかしながら、理論(計算化学)や実験での検証が進んだ結果、実際には図5のようなアルカリ金属イオンによる二酸化炭素分子の活性化とそれに続くMOPh-CO2 complex (仮に中間体Oとする)の形成が重要過程である可能性が高いことが示されています。
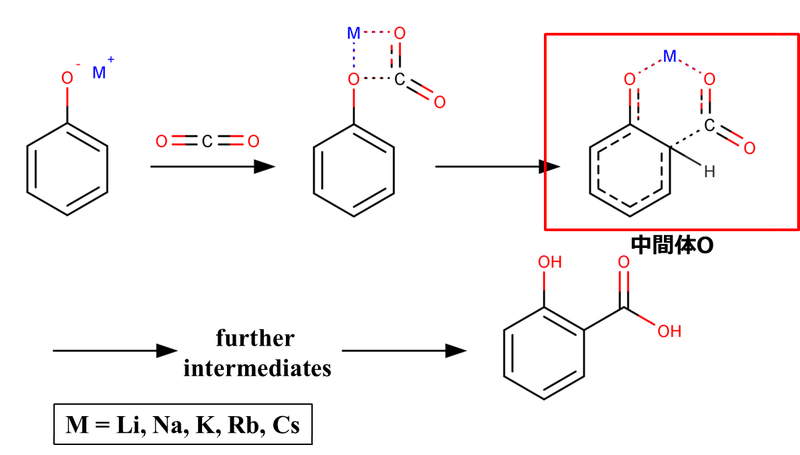
図5. アルカリ金属フェノキシドのオルト位カルボキシル化反応における第1段階および第2段階の中間体 [1-2].
このようにアルカリ金属イオンに配位することで二酸化炭素分子が活性化され、芳香環のオルト位に対する求電子攻撃が可能になります。因みにですが、このMOPh-CO2 complexは高圧下で他の二酸化炭素分子によって溶媒和されます。その点でこの反応における二酸化炭素は試薬であるとともに反応溶媒としても振る舞っていることになります。この反応の進行に高圧条件が必要な理由の一つかもしれません。
アルカリ金属イオンによって変化する反応位置選択性
さて、図5の反応中間体を見た時、勘のいい方ならアルカリ金属イオンのイオン半径の違いによって生成物のオルト:パラ非が変化する理由をなんとなく察せたのではないでしょうか。
図5ではオルト位で反応が進行する際の中間体Oが示されていますが、ではもしパラ位で反応が進行するとなった場合どのような中間体(仮に中間体Pとする)になるかを考えてみます。その場合、中間体Pは図6に示すような構造になる必要があると予想できます。
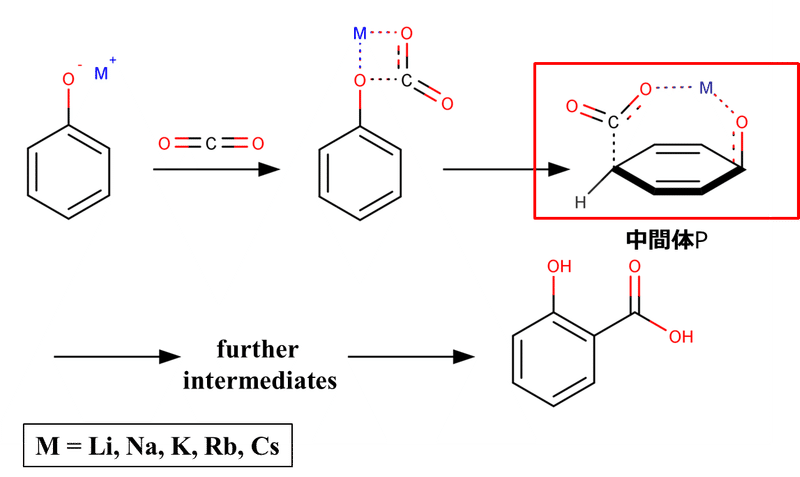
図6. アルカリ金属フェノキシドのパラ位カルボキシル化反応における第1段階および第2段階の中間体 [1-2].
すなわち、パラ位で反応が進行する際の中間体PはCO2-Mがベンゼン環をまたぐようなbridge構造になります。イオン半径が小さくO-M結合長が短いような時にこの中間体Pを成立させるためには、ベンゼン環を大きく変形させるような無理を強いることになってしまいます。そのため、中間体Pの形成は非常に不利になります。一方で、イオン半径が大きくなればそういった無理を強いることなくある程度余裕をもって中間体Pの形成が可能になります。このような理由によって、イオン半径が比較的小さいLi,Naではパラ体が生じず、イオン半径が比較的大きいK,Rb,Csではイオン半径の増加とともにパラ体の生成も増加していくという傾向を示すようになるのです。以上を踏まえると、アルカリ金属イオンがLi,Naの時のKolbe-Schmitt反応によるサリチル酸の合成は、反応がオルト位選択的というよりかはパラ位(もちろんメタ位も)非選択的といった方がより正確かもしれないですね。
ちなみに
上記の理由により、Kolbe-Schmitt反応によるサリチル酸の合成はリチウムフェノキシドかナトリウムフェノキシドで行うのが適切ということになるのですが、実はほぼ完全にオルト位選択的なのはリチウムフェノキシドだけでナトリウムフェノキシドはわずかながらパラ位でも反応が進行します。なので、リチウムフェノキシドを試薬として用いる方がいいんじゃないかと感じますが、まぁとはいえナトリウムフェノキシドのパラ位反応生成物の割合は微々たるものみたいですし[2]、試薬としての使いやすさがナトリウムフェノキシドの方が格段に上なのでナトリウムフェノキシドが使われているのだろうと思います。
まとめ
ナトリウムフェノキシドと二酸化炭素を用いたKolbe-Schmitt反応によるサリチル酸の合成において、反応がオルト位選択的なのはNaのイオン半径が比較的小さいためにパラ位で反応が進行する際の中間体構造を形成することが困難なためである。Naよりもイオン半径が小さいLiではさらにパラ位での反応が難しくなる。逆に、K,Rb,Csといったナトリウムよりもイオン半径が大きいアルカリ金属イオンのフェノキシドでは、パラ位で反応が進行する際の中間体構造を無理なく形成できるようになるため、パラ位での反応生成物の割合が増加していく。
参考文献
[1] Z. Markovic et al. J. Chem. Inf. Model. 2007, 47(4), 1520–1525.
[2] Z. Markovic et al. J. Chem. Inf. Model. 2006, 46(5), 1957-1964
この記事が気に入ったらサポートをしてみませんか?