AlphaGoの衝撃再び — タンパク質構造予測でAlphaFoldが今までのモデルに圧勝

原文記事: [阿尔法狗再下一城 | 蛋白结构预测AlphaFold大胜传统人类模型] (2018/12/03公開)
「研究したいタンパク質があるのだが、その構造と機能がわからない」 — これは分子細胞生物学の研究者が日々直面する最大の難題の一つである*a。アミノ酸配列測定技術が発展する中で、多くのタンパク質の配列がハイスループット*1に解析されているが、この配列決定の段階から実際に3次元構造を決定するまでの間には、未だに大きな距離がある。
生物の基本単位が細胞だとして、細胞の基本的な機能単位こそが、複雑多岐なタンパク質の1つ1つである。そしてまさにタンパク質の機能の本質を決定しているのが、タンパク質の構造である。タンパク質の機能を研究したり、それをターゲットとする薬剤を開発したいというとき、タンパク質の構造はとても重要な要素の一つになる。だからこそ、生物学には、構造生物学という学問領域までもが存在する。かの有名な中国人生物学者施一公(Shi Yigong*2)も、構造生物学の分野の第一人者の一人である。
2018年は、2年に一度開催される国際タンパク質構造予測コンテスト(CASP)の行なわれる年であった。このコンテストはすでに初開催から25年の歴史*3を迎えようとしていて、毎回世界中から数百ものスポンサーを集め、組織委員会によって選ばれたタンパク質*4の構造の定量的予測が競い合われる。この週末*3に丁度今回大会の結果が報告された。
報告の前に、大会組織委員会は参加者に向けて以下のような予告メールを送信した:
“CASP13 this year has observed unprecedented progress in the ability of computational methods to predict protein 3D structure. The reasons are not yet fully clear, but all this, including of course the results, will be discussed at the meeting.”
つまり、「今回の大会では今までにないレベルの進歩があった。その原因はよくわかっていないが、週末の学会の際に詳しく発表し、議論ができればと思っている」とのこと (CASPではコンテストの結果発表/統括/議論の場として4日間の学会が行われる)。
この”今までにないレベルの半端ない結果を出したチーム”は人々の興味を一気にそそりたてた。そして、今回の件の特殊性から、大会では学会への追加参加登録の枠を設け、その白熱を見届けたいというメディアに向けて追加でチケットを購入できるように措置をするほどだった。
アメリカ時間の金曜日(2018/11/30)早朝、今回大会の総合順位が正式に掲載された。A7Dという名前のチームが圧倒的にトップに君臨し、他のチームを遠く引き離しているという結果だった。その差は一体どの程度のものなのか。我々が過去数年分のコンテストの最終結果をまとめてみたところ、A7Dと今回の2位のチームの点数の差は、CASPが始まって以来約20年分の進歩の合計よりも高いというレベルだった*c。

縦軸: モデルの毎回の課題(=タンパク質)に対する予測近似性の成績累計。高いほど良い。
深い灰色は今年の97チームの成績で、浅い灰色は2016年の128チームの成績。赤い線がDeepMindのチーム、A7Dの成績。
# A7Dのモデルによるタンパク質構造予測の一例:
以下のいくつかの図で、(大会中は*4)構造未知のタンパク質に対する構造予測の各チームの結果が示されている。
縦軸: モデルによる予測と実際の構造の誤差をどれくらい許すか*6 (つまり線が右下に寄っているほど良いモデル)
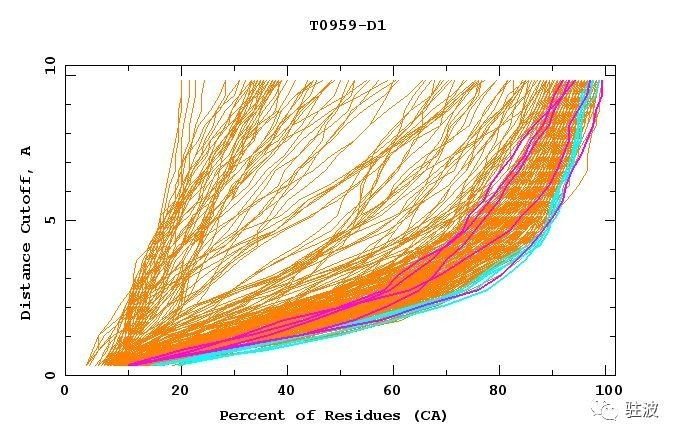
青色の線がA7Dによる予測結果で、ピンク色が総合順位2位のミシガン大学のチームによるもの。
他にもこれや
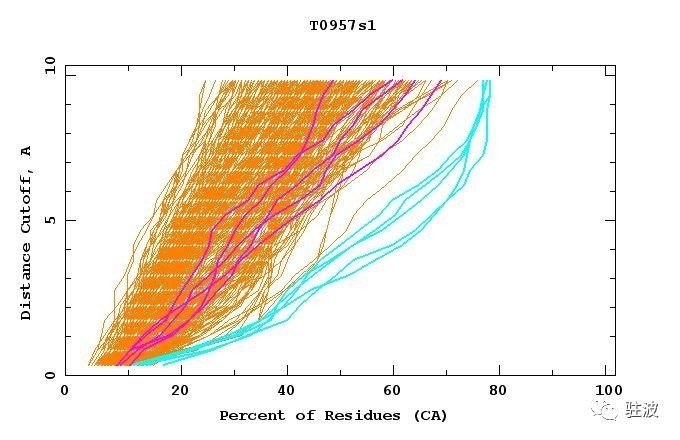
これも
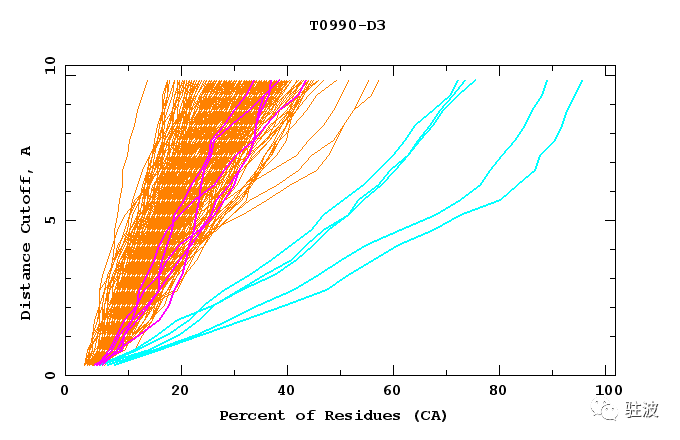
総合順位で1位になっただけでなく、DeepMindチームは対象となった43個のタンパク質のうち25個で成績トップとなっている; 比較として、総合2位のチームですら成績トップを取れたのは43個のうち3個だけである*7。
この圧倒的実力で優勝したやつらは一体どこの神がかったチームなのか? もうお分かりかもしれない、大会の後にA7Dは壇上で、彼らが実はDeepMindの研究員であることを明かした。そう、あのAlphaGoを開発したDeepMindである!報道によると、DeepMindはこのモデルをすでにAlphaFoldと名付けているそうだ*c。
実は、さかのぼること2017年10月、DeepMindはある公開インタビューで、人工知能の新薬開発への応用に興味を持っているということをすでに明かしていた。そして新薬開発への重要なステップが、ターゲットとなるタンパク質の立体構造の正確な予測計算である。このインタビューから丁度一年余りが過ぎた今、まさにDeepMindは世の中に、新たな分野での深層学習(ディープラーニング)の巨大なポテンシャルを証明したというわけである。
# DeepMindが今回挑んだ構造生物学
ある統計によれば、2010年の時点で、存在が認識されているタンパク質のうち立体構造が解き明かされているものはわずか0.6%しかないそうだ*c。この大きなギャップに立ち向かうべく、第一回のタンパク質構造予測コンテスト(Critical Assessment of Techniques for Protein Structure Prediction,CASP)が1994年にカルフォルニア州で開催された。解くべき問題をフォーマルに定義(=標準化)したことで、この20年余りで多くの計算モデルが開発された。筆者の指導教官であるChris Sander氏が、若かりし頃に理論物理学の分野から生物学に方向転換したのも、丁度この構造予測という生物学の新しい分野が出始めた頃であった。筆者自身もこの原稿を書くまで、Chrisが当時の第一回CASP大会の受賞者の一人であることは知らなかった。

筆者の指導教官Chris Sander氏。
“蛋白質”という漢字三文字をなぜか手書きできるドイツ人のお爺様。
歴史的に言えば、これらの計算モデルには主に3つの流派がある — Comparative Modelingという進化的アプローチ、threading mothodsという比較アプローチ、そしてfrom scratchというab initio (一から予測する)アプローチ。
進化的アプローチの核心となるアイデアは、タンパク質の進化の歴史的に同じ(もしくはほぼ同じ)起源を持つタンパク質を見つけて、それを出発点にして目標となる新たなタンパク質の構造を予測するというものである ; 比較アプローチの核心的アイデアは、進化的に同じ起源を持っていなくても、解析したいタンパク質のアミノ酸配列の断片を、今まで知られている配列と構造のリストと組み合わせて、新たに構造を予測するというもの ; そして、最も難しいが最も重要となるのがab initio流であり、この場合は全くもって類似するタンパク質が見つからないようなタンパク質の配列をゼロから予測するというものになる。ab initioは、ラテン語で”一から”、”最初から”の意味を持つ。
1999年、ab initio型のRosettaというモデルを、ワシントン大学のDavid Baker氏のグループが開発した。このモデルはMonte Carlo法と呼ばれる近似手法を用いて、長さおよそ100アミノ酸程度のいくつかのタンパク質の構造の予測に成功した。予測精度はRSMD(誤差の二乗平均)3.8オングストロームまで下がり、CASPIII (CASP第三回大会)の受賞チームの一つとなった*d。Baker氏は2003年に科学誌Scienceで、今度は長さ93アミノ酸の人工合成されたTOP7というタンパク質の構造予測に成功し、精度は誤差わずか1.2オングストロームまで差だったと報告した*e。2005年、BakerのグループはRosetta@homeという、スクリーンセーバープログラムを開発した。コンピュータが活動していないときにRosettaが構造解析のためのシミュレーション計算を勝手に行ってくれるというものである。このような”分散計算”的な手法で、数多くの人々の空き時間を計算資源として活用することで、大きな成功を収めた。
Baker Labが開発したスクリーンセーバープログラム、Rossetta@Home
近年では、CASP大会が継続して行われる中で、これらの流派の間の境目も少しずつ曖昧になってきていて、ますます多くの研究チームがこれらの三つのアプローチを全て一つに組み合わせて、より正確な一つの予測モデルを作り上げ始めている。ミシガン大学のYang Zhang氏のチームが開発したI-TASSERはそのようなチームの中の一つの成功例である。

UMichのYang Zhang教授とI-TASSER。このプログラムはすでに6000回以上引用されていて、141以上の国々で100,000人以上の研究者を助けてきた。
2008年にモデルが世に公開されてから、I-TASSERとその多くの亜種は、既に最も流行の構造計算モデルの一つとなり、ここ10年のCASP大会の上位常連である。今回のCASP大会では、Zhang氏のチームはI-TASSERとコンボリューショナルニューラルネットワーク(CNN)を組み合わせ、モデルの予測能をさらに一歩押し上げて、第2位を獲得した。
# AlphaGoのタンパク質構造予測モデルバージョンは、なぜ大成功を収めたのか
AlphaGoが世に出るだいぶ前から、CNNと強化学習(Reinforcement Learning)を用いて近似計算のステップ*6を実装を試してみた学者がいなかったわけではない*f。では、今回のAlphaFold*7はなぜ本大会で圧倒的な成績を収められたのか? AlphaFold*7の公式発表は特に出ていないため、我々は大会ホームページにある彼らの1ページのアブストラクト(概要)から探るにとどまった。


DeepMindがCASP大会のホームページで提出したモデルの概要
チームリーダーの一人であるAndrew Seniorによれば、今回DeepMindが提出した予測結果は三種類の深層学習由来のモデル(deep neural network)からなるとのこと。
モデル全体としては、一つの二次元ネットワークと、一つの点数評価ネットワークから構成されているとのこと。
二次元ネットワークでは、タンパク質の一次配列が、それぞれのアミノ酸の距離を予測するのに使われる。この計算の時点では、タンパク質の立体構造自体は特定できないが、このニューラルネットワークは、どのアミノ酸がアミノ酸の組が空間的に近い距離に存在するかを学習し予測することができる (=2次元のcontact matrixを作れる)ため、いわば一次元の配列情報を二次元の距離情報に変換している。
評価ネットワークへの入力データは、上記の1つ目のネットワークの出力に加えて、配列のマッチ度(Multiple Sequence Alignment,MSA)や幾何的構造 (Structure Genometry)の情報が含まれる。これらの情報がモデルに入力され、モデルの予測結果と実際の既知の構造の類似性を学習することで、最大パフォーマンス時の点数が学習される。予測のプロセスにおいては、この点数こそがモデルが最大化したい目標関数となる。*8
このニューラルネットワークに学習させるために、DeepMindは国際的なタンパク質データベース(PDB)の全ての既知のタンパク質構造を学習用データとして使った。個々のタンパク質を非常に多くのペプチド断片に分割し、それれの断片毎の構造の予測と評価を行う。これらの評価の結果が、伝統的なRossettaによる点数評価と共に、2つのネットワークにおけるパラメタの訓練に使われ、それに伴い目標関数が出力される。

CASP13中の一つの例、CASP13-T1008。モデルに学習させる過程をイメージした画像。
なるほどこれで完了か、と思ったら大間違い、DeepMindがすごいのはここからである。DeepMindは今回上で述べたモデルに、さらに一つ、いわば伝統的なfragmentation (まず計算モジュールを細かく分けて、のちにそれらの結果を統合する学習スタイル)を完全に無視する方法を取り入れている。このモデルにおいては、DeepMindはアミノ酸の間の結合角(torsion)を直接モデルの出力として、上述の二つのネットワークの出力する二次元的構造情報と全長評価に直接、勾配降下法(Gradient Descent, GD)を適用し、最終的に驚くほどの結果を叩き出している。このように、生物物理学(Biophysics)における伝統的な、特徴を個別に抽出していく方法を完全に無視した今回のモデルは、数年前のAlphaGoの新規性と実力を兼ね備えた姿を彷彿とさせる。
# 終わりに: 構造生物学の春がやってきたと言えるか
「DeepMind以外に、英国国内を含め数多くの研究員が種々の機械学習の方法を用いて同じ問題を解こうとしている」英国の科学者Liam McGuffin氏もそう言って楽観的な態度を示した。 「ここ数年でAIがこの分野で驚くほどの全身をもたらした。もしかしたら2020年頃には、タンパク質の構造予測の問題は根本的に解決されるかもしれない。私は非常にポジティブに感じている。」と言った。
構造生物学の領域では、今回の件が大きなブレイクスルーであることは疑いようがない。しかし、同時に多くの疑問や不安の声を読んでいるのも確かだ。実は、このモデルですら正確性がそこまで高いわけではなく、伝統的なモデルが正解できるのにこのモデルが思ったほどうまくいかないこともある。例えばCASP13-T0966-D1。これはE. Coli中のRRSPというタンパク質に対応し、Ras-Erk回路と相互作用する重要なタンパク質かつ、Rasに関連するガンを治療しうる薬剤ターゲット候補としても非常に重要なのだが、AlphaFoldはこのタンパク質の構造予測においては全体の平均レベルを下回っている。AlphaFoldのモデルはどのようなタンパク質の構造予測に関してより有効なのか?それらでなぜ有効なのか?これらの詳細はまだしっかり研究で明らかにされてはいない。このような曖昧なモデルを実用的に薬剤開発で使えるだろうか? まだ少し小さな疑問符が残るだろう。

DeepMindのモデルが失敗した例: タンパク質RRSP
青色の線がA7Dの結果を示し、ピンク色の線が今回総合第2位のミシガン大学のチーム
「モデルの正確性よりもむしろ、DeepMindが今回のモデルで周りが予測していたのに反して強化学習のアイデアを用いなかったことの方がむしろ興味深い。」そう語ったのはMITで人工知能の研究をしている博士課程の学生S氏。「DeepMindにはこの発想(強化学習)を試すべき1万もの理由があるが、最終的に公表されたモデルにおいてはそれは使われていなかった。DeepMindほどにリソースに溢れたチームですら使わなかったということは、強化学習のタンパク質構造決定問題に対する応用それ自体に対する警告と考えてもいいかもしれない。」と。
「さらに、モデルの中にはRosettaの点数評価が含まれている。」S氏は続けて語った。「DeepMindはこれらの伝統的な評価モデルを全て無視することを試したが、結局取り入れざるをえなかった。この事実から、長きにわたり研究者が知恵を絞ってきた伝統的アプローチも同じくらい重要といえるのではないか」と。
実は、生物学におけるAIの利用の例は他にも多くある。近年ではgoogleが主導する人工知能研究チームが生物/医薬開発の分野で全面的に力を発揮していて、ガンの医療診断画像からの判定、ゲノムの変異の検出、病気のリスク評価など様々な分野で、人間と同等のレベル、あるいは人間を超えるレベルの成績を叩き出している。しかし、これらの表面上では大成功に見えるようなモデルにおいても、モデルの普遍性、応用性、解釈可能性に関しては大きな障壁があることを忘れてはならない。
機械学習の応用のゴールに必要なのは、決して高精度のニューラルネットワークの活用だけではないだろう。より重要なのは、その分野において解決が急務とされている問題に関する深い理解、そして関連する分野のバックグラウンドを持つ人材の協力であろう。より多くの人がこの戦いに加わってくれれば、AI医療の未来は明るい。
*a Roy, A. et al., Nature Protocol, 2010より引用
*b 毎回のコンテストでの対象タンパク質は異なるのでもちろん一概に比較はできない。このような直接の比較は極めて不正確であることは理解しつつ、参考までに。
*c Guardian より
*d Simons et al., Proteins. 1999.
*e Kuhlman et al., Science. 2003.
*f Czibula et al., Int.J.Comp.Tech.Appl. 2011.
*1 High-throughput: 比較的短時間で大量に / 単位時間当たりの生産量が大きい、の意味。
*2 施一公 は中国清華大学生命科学院学長などを務め、中国国内で有数の有名な研究者。
*3 1994年初開催. 原文のニュアンスを少し変更 / オリジナル記事は2018/12/03公開
*4 CASP では、対象となるタンパク質の決定は主催者/参加者のどちらとも独立した組織により行われる、かつ、大会開始時には構造が実験的に解かれていない(が大会中には確実に解けるであろう)タンパク質或いは最近構造が実験的に解かれたばかりだが大会が終わるまで厳密に非公開となっているタンパク質をコンテストの対象として選ぶことで、平等性を保ちつつしっかり正答率評価ができるようになっているらしい。
*5
x軸 = 正しく位置をアサインできた残基の割合
y軸 = “正しい”と判定するためにどれくらいの誤差を許すか(オングストローム単位)
例えばx=70, y=5の場合、「5オングストロームくらいの誤差を許していいとすれば、70%は正しかったと言えるよ」という解釈。
*6 原文は”退火” = 焼きなまし / アニーリング だが、細かい話になるので近似計算 と意訳。
*7 原文AlphaGoになっていたが多分誤字(?)
*8 原文に忠実に行きましたがどこまで正確な記述なのか不明.. (アルゴリズムに関する記述全体)。 雰囲気を味わってくれればと思います。
トップ画はwikipedia Rosetta@home ページより転載.
https://en.wikipedia.org/wiki/Rosetta@home
文/袁博
校閲/范静萱、张涵雄、常亮
日本語訳 / 王青波
(2018/01/13 誤字を修正)