RDP classifierで微生物叢の解析を行う

2017年から2018年にかけて、メタバーコーディングによる土壌微生物叢の解析を行なっていたときに使っていたRDP classifierの使い方についてのメモ。
RDP classifierは、ある微生物叢の階層を視覚的に表現するのには向いているが、複数の微生物叢の構成を比較したい場合にはQiimeの方が向いているので、大量のサンプルを解析するようになってからは使わなくなった。
また、COPEreadはMac OS上では動かないので、PC上にインストールしたCentOSやスパコンで行っていた。
微生物叢サンプルからDNAを抽出し、16S PCR断片をV3-V4 プライマーセットで増幅、AMPure XPビーズで精製し、Nextera XT Index kit でIndex PCR、MiSeqでシーケンスした後のデータの解析。
① MiSeqのデータをダウンロードする。
以下の記事を参照。
② FastQOでクォリティーチェック
FastQCを以下のサイトからダウンロード。
http://www.bioinformatics.babraham.ac.uk/projects/download.html#fastqc
ショートカットの作成
$ vi .bash_profile
#edit it with path folder of fastqc and put the text below (also depend on your installation folder location)
PATH=$PATH:HOME/installation.folder.location/FastQC
#save it and load
$ source .bash_profile
$ fastqc -hOutputファイルの出力先を設定
$ mkdir fastqc.outputFastQCのラン
$ fastqc --nogroup --extract input_folder/*.fastq* -o fastqc.output/あるいは
$ fastqc -o sample_dir --nogroup --extract sample.fastq例)
$ mkdir 20170316_sample_dirmkdir 20170316_sample_dir
$ fastqc -o /Users/shigeru/Desktop/20170316_sample_dir --nogroup --extract /Users/shigeru/Desktop/20170310/170307_M01534_0233_000000000-AYKA5/*.fastq.gz(*はワイルドカード。これを使うことによって、フォルダ内の全ての.fastq.gzファイルを解析します)
*FastQCはMac上のアプリでも動作します。
③ 配列データのトリミング(トリミングはMac上で行ってもよいが、次のCOPEではLinuxが必要なので、サーバーに移してからトリミングを行う方が便利)
Trimmomaticを以下のサイトからダウンロードします。
http://www.usadellab.org/cms/?page=trimmomatic
シートカットの作成
$ vi .bashrc
#edit in text editor and paste the text below (also depend on your installation folder location)
alias trimmomatic=’java-jar $HOME/folder.install.location/Trimmomatic-0.36/trimmomatic-0.36.jar’
#save and load
$ source .bashrc
$ trimmomatic -hTrimmomaticでPair-endのトリミング
$ mkdir trim.output
$ trimmomatic PE -threads 6 input_folder/E0-1_S16_L001_R1_001.fastq input_folder/E0-1_S16_L001_R2_001.fastq trim.output /E0-1_R1_trim.fastq trim.output /E0-1_R1_up.fastq trim.output /E0-1_R2_trim.fastq trim.output /E0-1_R2_up.fastq ILLUMINACLIP:/home/inysumerta/tool/Trimmomatic-0.36/adapters/Truseq-PE.fa:2:30:10 LEADING:10 TRAILING:10 SLIDINGWINDOW:4:15 MINLEN:75 >& output_folder/E0-1.stats例)ショートカットを使わずにやってみた例。特にスパコンなどで管理者以外がjavaでtrimmomaticをランさせる場合、pathがうまく通せないことがあるので、上記でうまくいかないときには下記のように入力してみるとよい。
$ java -jar /Users/shigeru/data/Trimmomatic/Trimmomatic-0.36/trimmomatic-0.36.jar PE -threads 6 -phred33 01-Nov-1_S46_L001_R1_001.fastq.gz 01-Nov-1_S46_L001_R2_001.fastq.gz 01-Nov-1_S46_L001_R1_001.trim.fastq unpaired.01-Nov-1_S46_L001_R1_001.trim.fastq 01-Nov-1_S46_L001_R2_001.trim.fastq unpaired.01-Nov-1_S46_L001_R2_001.trim.fastq ILLUMINACLIP:NexteraPE-PE.fa:2:30:10 LEADING:10 TRAILING:10 SLIDINGWINDOW:4:15 MINLEN:75 >& 01-Nov-1_S46_L001.statsILLUMINACLIPのオプションに注意。
Result: E0-1_R1_trim.fastq; E0-1_R1_up.fastq; E0-1_R2_trim.fastq; E0-1_R2_up.fastq; E0-1.stats
④ COPEread(COPEreadはMac上では動かないので、Linuxサーバーを利用する)
COPE: An accurate k-mer based pair-end reads connection tool to facilitate genome assembly. Binghang Liu; Jianying Yuan; Siu-Ming Yiu; Zhenyu Li; Yinlong Xie; Yanxiang Chen; Yujian Shi; Hao Zhang; Yingrui Li; Tak-Wah Lam; Ruibang Luo. Bioinformatics (2012) 28(22): 2870-2874; doi: 10.1093/bioinformatics/bts563
以下のサイトからCOPEreadをダウンロードします。 https://sourceforge.net/projects/coperead/
COPEreadインストール
$ tar xvf [ダウンロードしたファイル]
$ make
$ sudo make installCOPEreadのラン
$ cope -a trim.output /E0-1_R1_trim.fastq -b trim.output/E0-1_R2_trim.fastq -o COPE/E0-1 -m 0 -u 150 -l 10 -s 33 > E0.1.logResult: E0-1.connect.fq; E0-1.unConnect_1.fq; E0-1.unConnect_2.fq; E0.1.log
⑤ fasta fileを作成(fastqファイルをfastaファイルに変換)
FastX-toolkitをダウンロードする。http://hannonlab.cshl.edu/fastx_toolkit/
解凍してインストールするだけで使用可能。
$ tar jxvf fastx_toolkit_0.0.13_binaries_Linux_2.6_amd64.tar.bz2インストールされるbinにPATHを通す(あるいは、PATHの通っている場所へ移動する。
fastq to fastaをランする。
$ fastq_to_fasta -i COPE/E0-1.connect.fq -o FastX.output/E0-1.connect.fasta -Q 33Result: E0-1.connect.fasta
注意事項:FASTX-Toolkitの各種ツールを実行するときは、オプションで -Q 33 を与えることを忘れないこと。
・fastq_to_fasta
fastqファイルに含まれる各リードをfastaに変換します。単純に考えれば、fastqファイルの3行目と4行目を削除したものになります。また、Nが一つでもあるリードは除去されてなくなってしまいます。その代り、どんなにクオリティ値が低いリードでも除去されません。
【optionの説明】
-h: ヘルプを表示します。
-r: fastqファイルの各リード1行目のシーケンスIDをただの番号に変えます。ファイルの先頭から各リードに1から順番に番号が付きます。
-n: デフォルトではNが一つでもあるリードは除去されます。このオプションを入れると、Nがあっても除去されません。
-v: 処理前後でのリード数などを出力してくれます。
-i: 入力ファイルを指定します。
-o: 出力ファイルを指定します。指定しない場合は標準出力に出力されます。
【実行例1: 普通に変換】
$ fastq_to_fasta -i test.fastq -o test.fasta -Q 33
【実行例2: 番号付出力、及びNがあっても除去しない】
$ fastq_to_fasta -i test.fastq -o test.fasta -r -n -Q 33
test.fastqからtest.fastaへの変換はawkコマンドを使う方法もあります。
$ awk '(NR - 1) % 4 < 2' test.fq | sed 's/@/>/' > test.fa
⑥ RDPclassifier
RDPclassifierをダウンロードします。
Download:https://sourceforge.net/projects/rdp-classifier/
解凍してインストール。
$ unzip rdp_classifier_2.12.zipjavaでRDPをランする
$ java -Xmx1g -jar tool/rdp_classifier_2.12/dist/classifier.jar -o RDP.output/E0-1.classified.txt -h RDP.output/E0-1.hier.txt FAstX.output/E0-1.connect.fastaResult: E0-1.classified.txt; cnadjusted_E0-1-hier.txt; E0-1-hier.txt
このテキストファイルを開くと、微生物叢の数値データが得られる。
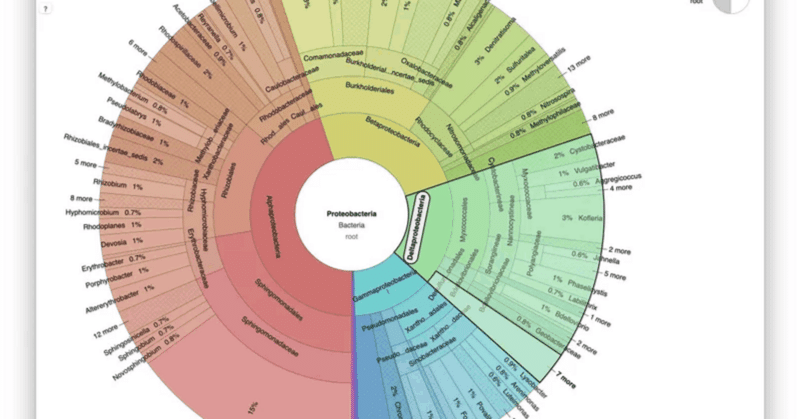
⑦ Kronaで視覚化
Kronaをgithubからダウンロードしてインストールします。
$ git clone https://github.com/marbl/Krona.git
$ sudo ./install.pl
$ ktImport Kronaをランする。
$ cd Kronaのフォルダーの中へ移動
$ ktImportRDP E0-1.classified.txt –o E0-1.rdp.Kronal.htmlResult: rdp.krona.html
このhtmlファイルを開くと、図に視覚的に表示されます。

Bacteriaをクリックすると、下の階層のデータが視覚化されます。

Bacteria > Probacteria までクリックして表示した例。

この記事が気に入ったらサポートをしてみませんか?