バイオインフォマティクスのすすめ(4)

第4回目の今回は,配列アライメントがどういう手順(アルゴリズム)で計算され結果が出力されるのかを解説していきます.
目次
0.始めに
1.配列アライメントの種類とアルゴリズム
2.グローバル・アライメント
3.ローカル・アライメント
4.Dot Plotの作成
5.マルチプル・アライメントの原理
6.まとめ
0.始めに
生命科学の研究現場では,新しいテーマ,新しいターゲットを手にしたり,学会用のポスター・スライド作成や論文作成で配列アライメントを提示するよう求められます.しかし,ユーザーが実際に手を動かすのは配列アライメントを行うために,サーバー上で配列を入力するだけで,あとはサーバー側が勝手に計算を行ってくれるので,実際にどういう原理で配列アライメントが出力されるのかを知らない・興味ない人が多いかもしれません.
この章を読み進めることによって,配列アライメントの実際とそれが出力された後に,我々,末端ユーザーがしなければならないこと,常に念頭に置かなければならないことを把握できるようになれば幸いです.
1.配列アライメントの種類とアルゴリズム
① 配列アライメントの種類
まず配列アライメントには,2種類あります.
ペアワイズ・アライメント
マルチプル・アライメント
名前の通りですが,ペアワイズ・アライメントは2本の配列を整列させる配列アライメントです.一方,マルチプル・アライメントは,3本以上の配列アライメントです.配列アライメントの基本であるペアワイズ・アライメントについて見ていきましょう.
もし,あなたが2本の短い配列を,目を使って重ね合わせようとしたら何をしますか?

第3回目で紹介したこれらは,私も実際に目を使って重ね合わせました.
多くの人がそうすると思いますが,いきなり異なる文字を探すのではなく,まずは同じ文字を検索します.上下に同じ文字を配置して(今回は字を赤くし)最後に残った黒字を整列させます.
しかし,このような目で行うアライメントは配列数が長くなればなるほど,比較配列の同異に対して一貫性を維持することが難しくなります.この問題を解決するために,比較文字の一致(マッチ)・不一致(ミスマッチ)・ギャップそれぞれにスコア付けを行います.当然,マッチスコアは点数が高く(正の値)ミスマッチスコアは点数が低く(負の値)なります.またギャップはペナルティとしてギャップの長さ分の負の値が加算されます.ペアワイズ・アライメントのスコア例を以下に示します.もちろん,これが全てではなくプログラムによって多少の違いがあります.
マッチとミスマッチ
マッチ:正の値(例:+1)
ミスマッチ:負の値(例:-3)
ギャップペナルティ
-4g(gはギャップの長さ)
ペアワイズ・アライメントのスコアはDNA配列とアミノ酸配列でも異なります.DNA配列の塩基置換・欠失・挿入はDNA自身の変異にのみ影響されるため,マッチとミスマッチのスコアは原則,上のコラム通りになります.
一方,アミノ酸配列の場合,アミノ酸残基の置換・欠失・挿入は,セントラルドグマ上位のDNA塩基の変異によって影響され,その変異が進化の過程において,いつ,どの段階で起きたのかを考慮しなくてはなりません.そのため単純に残基の同異をスコア付けすることができません.アミノ酸配列のアライメントでは,BLOSUMや PAMというスコア付けのためのアルゴリズムを利用するのが一般的です.
基本的にソフトウェア側に組み込まれているので,我々ユーザーは気にする必要はありませんが,BLOSUM を用いて出力した配列アライメントと PAM を用いて出力した配列アライメントでは結果が異なるのは言うまでもありません.
② アルゴリズムの種類
配列アライメントのスコアが決定したので,次はどのような手順(アルゴリズム)で配列の一致・不一致・ギャップを決定しているかを見ていきます.
ここでは配列アライメントのアルゴリズムは2種類あることを押さえていてください.
グローバル・アライメント
ローカル・アライメント
次章から,各アルゴリズムについて簡単に解説します.
2.グローバル・アライメント
ここから先はDNA配列の配列アライメントについて見ていきます.
グローバル・アライメントが何かを勉強していく前に,アライメントのアルゴリズムを勉強する上で基本となる考え方を示します(下図).
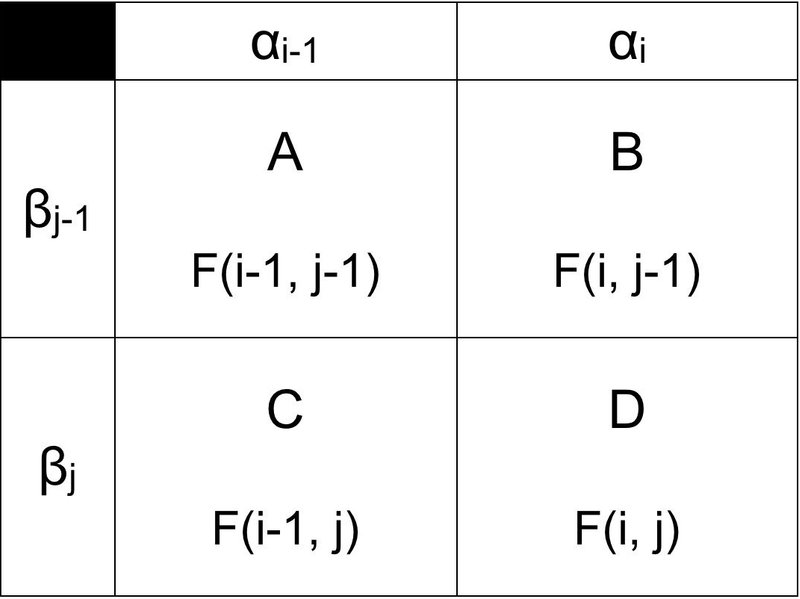
たとえどんなに長い配列でもこれが基本形になります.2本の配列 α と β について配列比較をするとき,配列 α の i-1 番目と i 番目,配列 β の j-1 番目と j 番目(ただし i と j は自然数であること)を比較します.上図の D を終点として,どの道のりを行くかでスコア付けが行われます.すなわち,ルートは「A → D」「B → D」「C → D」の3候補あり,D のマスのスコアを関数 F(i, j) とすると,他の3マスも同様に関数 F で表すことができます.
「A → B」や「A → C」のルートも候補にならないか疑問に思う人もいるかもしれませんが,上図は一般化しているので,マスの平行移動で「C → D」や「B → D」に対応できます.
① まずは,ルート「A → D」について考えてみましょう.
「A → D」は最も基本的なルートです.「α(i-1) と β(j-1) のマス」から右下の「αi と βj のマス」を見るとき,αi と βj の文字が一致していれば加算(+1)不一致の場合は減算(-3)されます.この加算・減算を関数 +s(Xi, Yj) と置くことにします.
② 次に,ルート「B → D」と「C → D」について考えてみましょう.
「B → D」と「C → D」は縦横の違いはありますが,考え方は同じなので一度に説明します.そもそも「B → D」と「C → D」はどういう状況を考慮しているか分かりますか?
上図の配列 α, β と塩基番号に注目してください.「縦に移動する」「横に移動する」ということは,それぞれ「i 番目の α を維持しつつ β を j-1 番目から j 番目に移動する」「j 番目の β を維持しつつ α を i-1 番目から i 番目に移動する」ことであり,実はこれがまさにギャップを示しています.そのため,ルート「B → D」と「C → D」は両者とも減算(-4)されます.
①と②を下図で纏めます.スペース上,A~Dを省略しました.
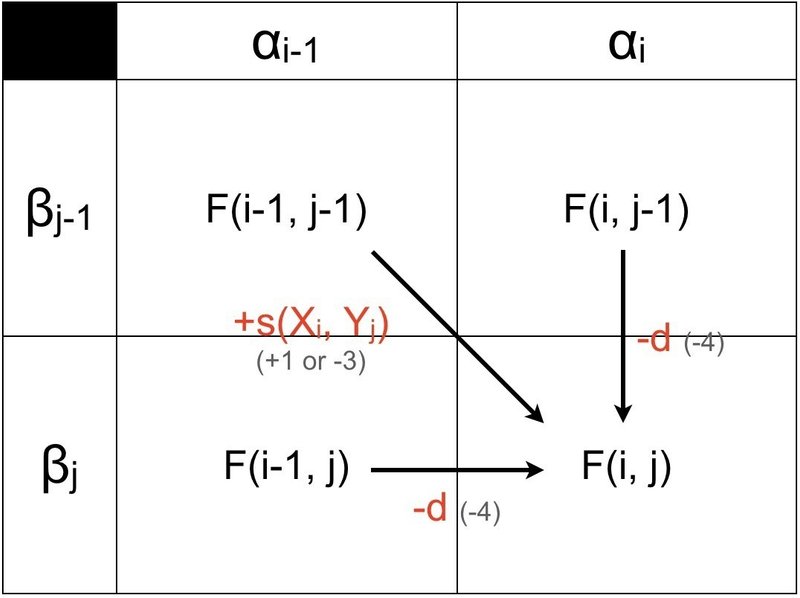
グローバル・アライメントの場合,Dへ通ずるルートのうち,最大値を選び出し,どの経路を利用したかを記録します.これを一つの式で示すと下のようになります.
グローバル・アライメントのスコアF(i, j)の計算方法
F(i, j) = max{
F(i-1, j-1) + s(Xi, Yj), A → D
F(i, j-1) - d, B → D
F(i-1, j) - d, C → D
}
数式だけ見てもよく分からないと思いますので,以下にグローバル・アライメントを用いた例を示します.4塩基の重ね合わせなので目で見た方が早いですがご了承ください.
① (i, j) = (1, 1) のとき,つまり,(i-1, j-1) = (0, 0) のスコアを 0 とします.
② α と β の配列で同じ文字のマスの背景を塗りつぶします.
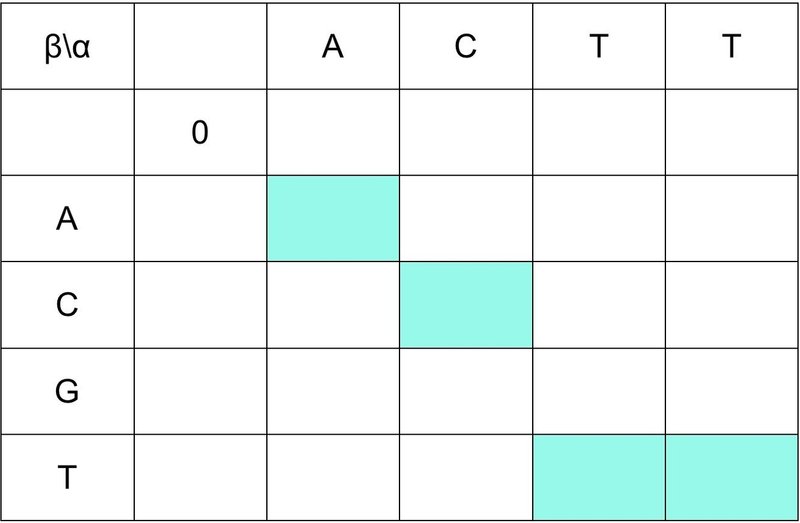
③ (0, j+n) と (i+n, 0) はギャップとして,-4ずつ減算していきます.
④ F(0, 0) = 0を起点としてななめ方向は+1ずつ加算していきます.
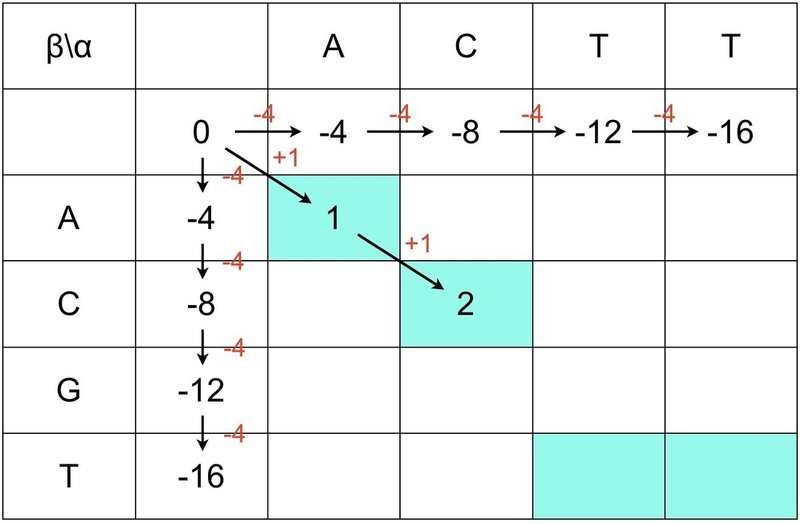
④ 背景が白いところは下図のように減算していきます.
⑤ このときF(i, j)は常に最大値を引くことを忘れないでください.
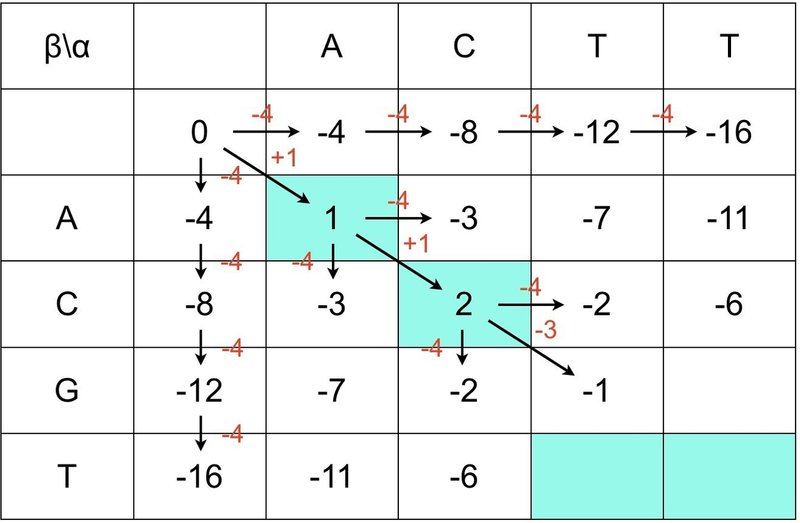
⑥ 最後に全てのマスをスコア(最大値)で埋めましょう.
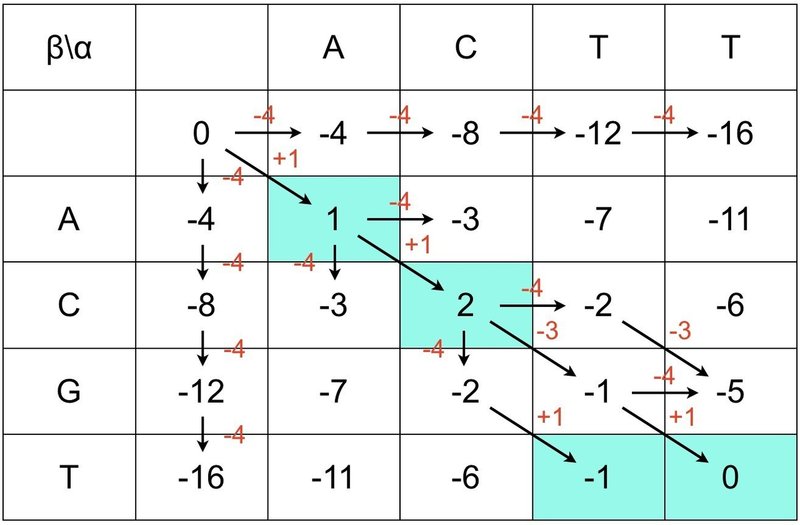
⑦ 右下から左上に向かってスコアの高いところを順に一筆書きします.
⑧ これでグローバル・アライメントが完成しました.

G と T 以外が赤字で表示されるだろうというのは想像に難くないですね.今回はギャップがなかったですが,ギャップがある場合は,赤矢印も左へ上へ移動します.
3.ローカル・アライメント
ローカル・アライメントの場合もグローバル・アライメントと同様に,Dへ通ずるルートのうち,最大値を選び出し,どの経路を利用したかを記録します.しかし,1点だけグローバル・アライメントとは異なる条件があります.それを以下に数式で示します.
ローカル・アライメントのスコアF(i, j)の計算方法
F(i, j) = max{
0,
F(i-1, j-1) + s(Xi, Yj), A → D
F(i, j-1) - d, B → D
F(i-1, j) - d, C → D
}
上式の通り,"0" が含まれています.これは,アライメントのスコア付けの結果が全てのマスにおいて,0 を下回る(負になる)ことは無い,ということを示しています.
グローバル・アライメントでは,例えば,ギャップペナルティは -4g なので,右か下に行くほど負の値が大きくなっていきましたが,それらが基本 0 になると考えると良いでしょう.
それでは,グローバル・アライメントと同様に,ローカル・アライメントの具体例を以下に示します.比較のために全く同じ配列を用います.こちらも配列が短いので目で見た方が早いと思いますが,グローバル・アライメントとどう異なっているのかを知るためにも良い機会であると思います.
最初はグローバル・アライメントと同じです.
① (i, j) = (1, 1) のとき,つまり,(i-1, j-1) = (0, 0) のスコアを 0 とします.
② α と β の配列で同じ文字のマスの背景を塗りつぶします.
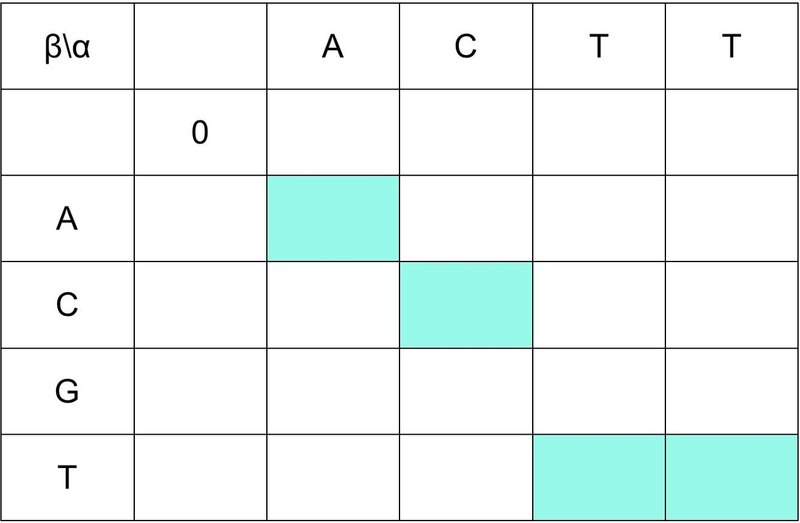
③ グローバル・アライメントと同様にスコア付けを行います.
④ スコアが負の値になると,最大値 0 が引かれることに注意します.
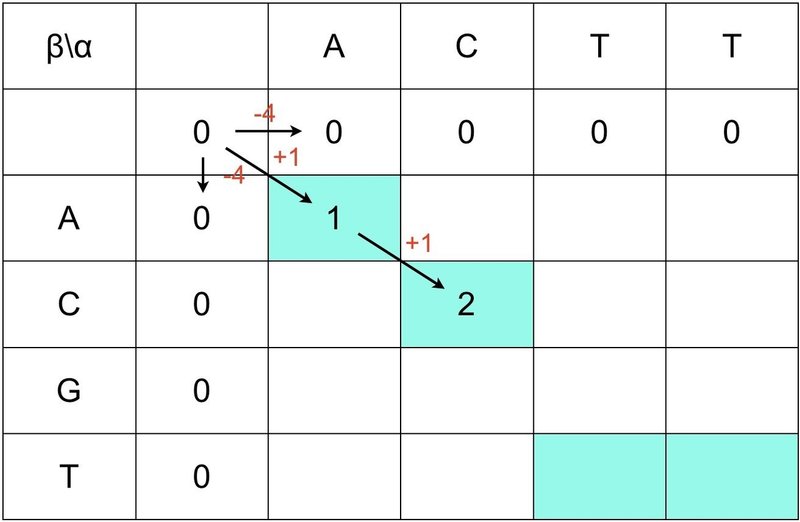
⑤ 最後に全てのマスをスコア(最大値)で埋めましょう.
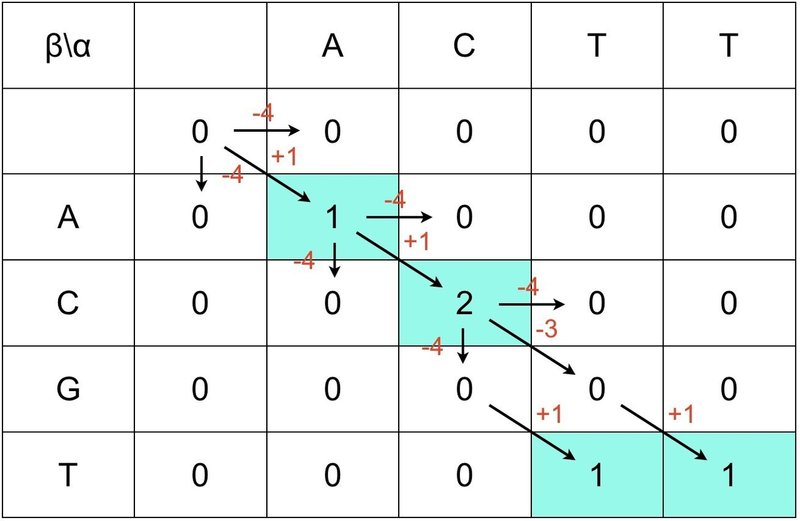
⑥ 最高点から0に向かって矢印を引きます.
⑦ これでローカル・アライメントが完成しました.
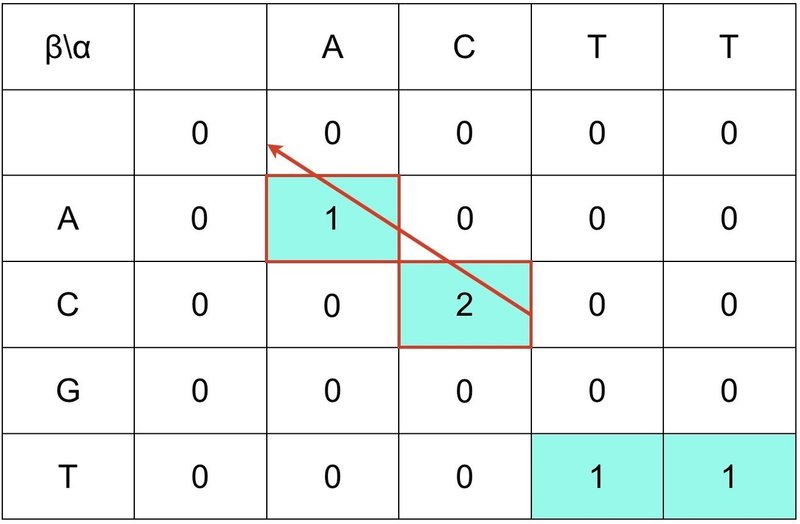
得られたローカル・アライメントは 1番目と 2番目の AC になります.
グローバル・アライメントとローカル・アライメントについて,駆け足で説明してきましたが,何となく違いが分かったでしょうか?ローカル・アライメントの方がすっきりしていると思った方も多いのではないでしょうか?
実際,配列全体を比較して配列の末尾から先頭まで一筆書きをしなくてはならないグローバル・アライメントは計算量も多く時間もかかります.配列アライメントのメリットは2本,もしくは3本以上の指定した配列を比較するのみならず,不特定多数の配列の中から似ている(進化的に近い・相同な)配列を探し出してくることができるということです.この場合は圧倒的にローカル・アライメントに軍配が上がります.これについては次回,説明します.
4.Dot Plotの作成
これまでの説明で,配列アライメントによって,どの文字が一致するか・しないか,どの領域が Indel であるかを推定できることを示してきました.ここで紹介する Dot Plot とは,長いDNA配列の配列比較を視覚化するツールです.今回は,YASS : : genomic similarity search tool for nucleic sequences in fasta format というサーバーを用います.
Googleで上のキーワードを検索するか下記リンクをクリックしてください.
http://bioinfo.lifl.fr/yass/index.php
上のタブの web server のページに必要事項を入力して最後に run YASS をクリックするだけです.必要事項も配列だけなので難しくないと思います.
以下に,EEF2 と EFT2 の比較結果と EEF2 と PH1899 の結果を示します.
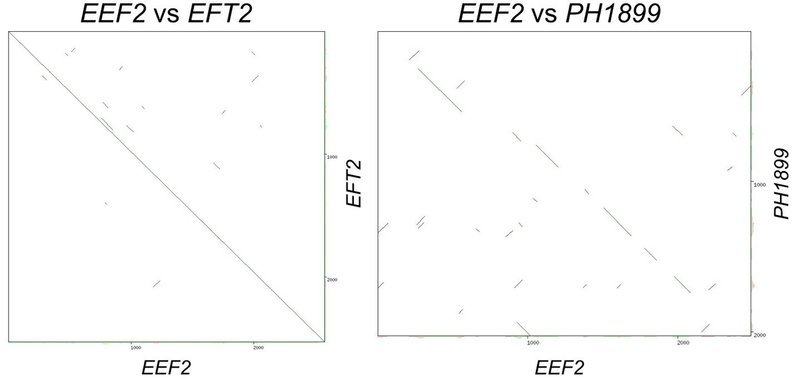
左の図は EEF2(Human の翻訳伸長因子 eEF-2)と EFT2(Yeast の eEF-2)のDNA配列比較を視覚化した結果です.Human も Yeast も同じ真核生物であるため,DNA配列長も配列類似性もそこまで大きく異なりません.一方で,右の図の EEF2 と PH1899(好高熱性古細菌 Pyrococcus horikoshii の翻訳伸長因子 aEF-2)との配列比較では,我々,真核生物の祖先にあたる古細菌に比べて,ヒトの eEF-2 は,実際には100 アミノ酸残基,300 DNA 塩基ほど長いですが,保存性の高い領域も存在することが示されています.
5.マルチプル・アライメントの原理
最後に,3本以上の配列を比較するマルチプル・アライメントについて原理と実情を示します.
始めに,3本以上の配列を同時にアライメントするのは現実的ではありません.グローバル・アライメント,ローカル・アライメント共々,マスを用いて説明しましたが,2本の配列でさえ,計 i × j マスの計算をする必要がありました.それが 3本になると i × j × k となり 4本では i × j × k × l と,配列の種類が増えれば増えるほど,x^(n)(x: 配列の長さ,n: 配列数)で計算量が増えていくことになります.
ではどうするかというと,類似性の高い配列のペアワイズ・アライメントを行い,次に類似性の高い配列とさらにアライメントを行うという手法を取っています(下図).
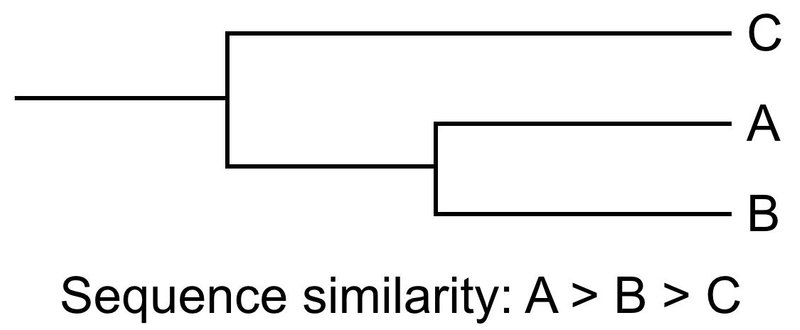
このとき,以下のように,A と B でアライメントをして,C と D でアライメントをする方が効率的に見えるかもしれません.
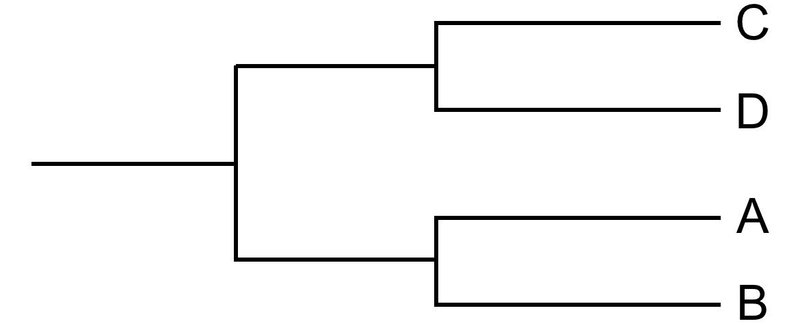
しかし,先述の通り,配列アライメントはただ重ねて整列させるだけではないので,もし,それぞれのペアワイズ・アライメントを行ってしまうと同異の判定基準が複雑になるため,これも現実的ではありません.
マルチプル・アライメントでは,2つ上の図のように重ね合わせていきますが,ここでも問題が生じます.それは C と比較する配列です.折角,A と B を重ね合わせてアライメントスコアを算出しても,次に何と C を重ね合わせてアライメントスコアを決定すれば良いのでしょうか?
こればかりは,私も単語を知っているだけですが,C 以降の配列との重ね合わせは直前で算出される「配列対のアライメントスコア」を用いるようです.この配列対のアライメントスコアに対しても定義づけが必要です.
6.まとめ
今回は内容が濃かったこともあり,冗長的になってしまいましたが,いかがでしたでしょうか?配列アライメントを利用する側としては,結局のところ,使えればそれでいいのかもしれません.
これは自身の実体験なのですが,当時の上司から配列アライメントの出力と,実際の構造(予測モデルも含む)を見ながら,配列アライメントの書き換え(実際にIllustratorでカチカチやりました)をせよ,と命ぜられたことがありました.当時の私は研究室に配属したばかりの学部4年生で,「何故,書き換える必要があるのか?」みたいな返しをしたら,配列アライメントは所詮,計算結果だから信用できない等と色々言われたことを,今でも何となく記憶に残っています.
そしてその1年後に履修した特論で,何となく上の会話が解決することになります.
このシリーズの中でもよく話題に上っていますが,計算の不完全さはある程度は許容すべきで,だからこそ私は,末端ユーザーはそれらを取捨選択する権利を持つと考えています.そして計算屋は我々が取捨選択してもいいように数多のアルゴリズムや統計量を定義していると期待しています.
次回からは,今回解説したローカル・アライメントを用いた遺伝子配列の検索,遺伝子領域の予測手法について紹介していきたいと考えています.
それでは,今後ともどうぞよろしくお願いいたします.
この記事が気に入ったらサポートをしてみませんか?