バイオインフォマティクスのすすめ(1)

この第1回目から配列比較(配列アライメント)について行っていきます.
今回は配列アライメントのための準備(配列の取得)について記します.
目次
0.始めに
1.配列情報の取得
2.ファイル名の付け方
3.ファイルの中身(FASTA形式について)
4.まとめ
0.始めに
この回では実際に配列アライメントをしてみます.DNA配列とアミノ酸配列の両方とも行います.
最初に手順を示します.
① 配列情報の取得(KEGG GENES DATABASE)
⬇️
② 配列アライメント(NPS@)
1.配列情報の取得
最終目標は配列アライメントですが,当然比較したい配列が必要です.そこで,まずは配列を検索して取り出します.配列検索はKEGG GENES DATABASEを用います.もちろん,配列検索データベースは他に幾つもあるので使い慣れているデータベースがあればそちらをお使いください.
GoogleでKEGGと検索する,もしくは以下のURLを打ち込みます.
https://www.genome.jp/kegg/genes.html
KEGG: Kyoto Encyclopedia of Genes and Genomes
生物種を特定しないでGENEを検索することもできますが,生物種(Organism)を指定してGENEを検索することができます.
ここでは,生物種およびGENE両方とも指定します.GENE枠にはタンパク質名(慣用名・正式名)を入力することもできます.
Search Organism( "bsu" )for ( "ftsz" ) と入力し,Goを押します.
※ ""は除く
Bacillus subtilis (Bsu) のftszは1項目しか出ないはずなので,そのままリンクをクリックすると,以下のページに飛びます.
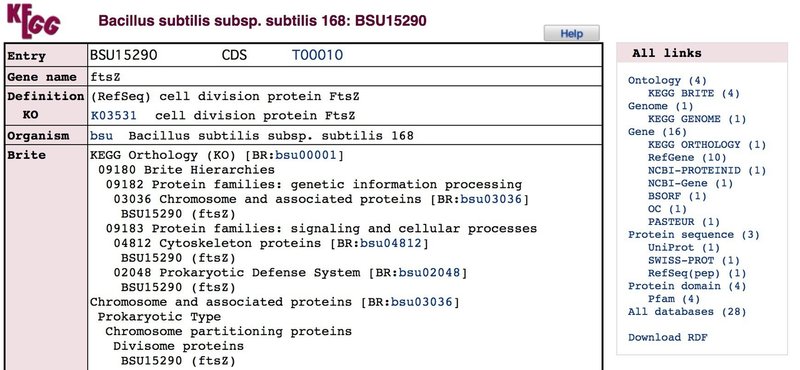
このページを下までスクロールすると,以下のようにDNA配列とアミノ酸配列情報が記されています.

これらの配列情報をユーザーが使用しているPCのOSに標準装備されているテキストエディタやTerminalに上図のように選択しコピペして保存します.保存形式は ".txt" や ".rtf" でも良いですが,今後の事を考えると拡張子変更をして ".seq" で保存するのが良いのかな?とも思います.
次に,Bsuのftszと比較するために,Staphylococcus aureus (Sau) のftszを用います.
Bsuのftszと同様の方法で検索します.
Search Organism( "sau" )for ( "ftsz" ) と入力し,Goを押します.
※ ""は除く
Staphylococcus aureusのftszも1項目しか出ないはずなので,そのままリンクをクリックすると,以下のページに飛びます.
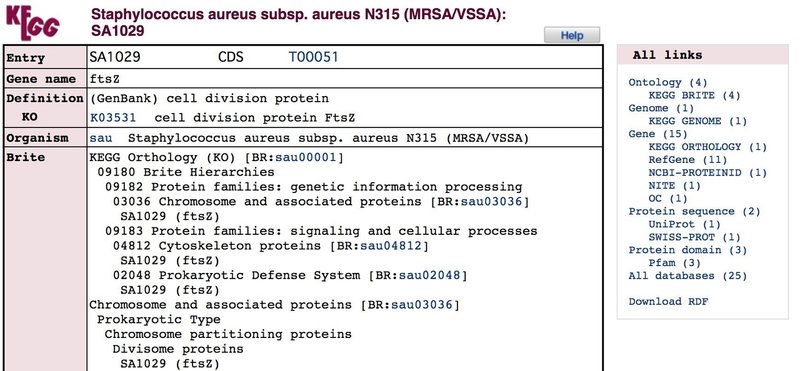
同様に,このページを下までスクロールするとDNA配列とアミノ酸配列情報が記されているので,それぞれを別々のテキストエディタにコピペします.
2.配列の保存方法(FASTA形式について)
DNA配列やアミノ酸配列ともに,配列(DNAの場合4種類,アミノ酸の場合20+α種類のアルファベット文字列)だけをテキストエディタに記して保存する方法と,これから説明するFASTA形式(FASTAフォーマットともいう)という記述式を用いて保存する方法があります.
記述方法に強制力は無いのでどちらでも構いません.しかしながら,特に複数の配列を同じファイルに保存する場合など,配列のみを記すと後で見返したときに不都合になりかねない,というのは想像に難くないと思います.
ですので,FASTA形式でまとめる方ことをお勧めします.様々なサーバー,特にシーケンスアライメントのソフトウェアの多くでは,FASTA形式が好まれ,世界標準化しているので,FASTA形式を覚えておく必要があります.
そもそもFASTAとは,シーケンスアライメントのソフトウェアパッケージの一つです.現在は,FASTAはほとんど使われず,後述するBLASTの方がメジャーです.
では,FASTA形式とは何かについて簡潔に解説していきます.簡単に言えば下のコラムの通りですが…
FASTA形式の例
(1番目はbsu_ftszのアミノ酸配列,2番目はsau_ftszのアミノ酸配列)
>bsu_ftsz_aa
MLEFETNIDGLASIKVIGVGGGGNNAVNRMIENEVQGVEYIAVNTDAQALNLSKAEVKMQIGAKLTRGLGAGANPEVGKKAAEESKEQIEEALKGADMVFVTAGMGGGTGTGAAPVIAQIAKDLGALTVGVVTRPFTFEGRKRQLQAAGGISAMKEAVDTLIVIPNDRILEIVDKNTPMLEAFREADNVLRQGVQGISDLIATPGLINLDFADVKTIMSNKGSALMGIGIATGENRAAEAAKKAISSPLLEAAIDGAQGVLMNITGGTNLSLYEVQEAADIVASASDQDVNMIFGSVINENLKDEIVVTVIATGFIEQEKDVTKPQRPSLNQSIKTHNQSVPKREPKREEPQQQNTVSRHTSQPADDTLDIPTFLRNRNKRG
>sau_ftsz_aa
MLEFEQGFNHLATLKVIGVGGGGNNAVNRMIDHGMNNVEFIAINTDGQALNLSKAESKIQIGEKLTRGLGAGANPEIGKKAAEESREQIEDAIQGADMVFVTSGMGGGTGTGAAPVVAKIAKEMGALTVGVVTRPFSFEGRKRQTQAAAGVEAMKAAVDTLIVIPNDRLLDIVDKSTPMMEAFKEADNVLRQGVQGISDLIAVSGEVNLDFADVKTIMSNQGSALMGIGVSSGENRAVEAAKKAISSPLLETSIVGAQGVLMNITGGESLSLFEAQEAADIVQDAADEDVNMIFGTVINPELQDEIVVTVIATGFDDKPTSHGRKSGSTGFGTSVNTSSNATSKDESFTSNSSNAQATDSVSERTHTTKEDDIPSFIRNREERRSRRTRR
FASTA形式は,1つのシーケンスデータ(アルファベット文字の羅列)をプレーンテキスト(文字コードのみで構成されるテキストデータ)を用いて記述する方法です.難しくありません.
① 1行目の先頭に ">" を記述し,ヘッダ行とします.
ヘッダ行は1行目のみで,>以降,スペースをあけずにヘッダを入力
(正)>bsu_ftsz_aa (誤)> bsu_ftsz_aa
② 2行目以降は,実際のシーケンスデータが記されます.
このシーケンスデータは改行を認識しません.
③ 複数種類の配列を羅列する場合,次に ">" で始まるヘッダ行の登場で
シーケンスデータが区切られます.
つまり,2番目以降の配列の前行に ">"行を置かないと,2番目の配列
も1番目の配列に含まれると認識されてしまいます.
3.ファイル名の付け方
最後に,ファイル名の付け方について,言及しておきます.
これはバイオインフォマティクスに限らず,どの場面でも重要になってくることですが,もし今後,この作成したファイルを自分以外の誰かに見せる・引き継ぐことが一切無いと断言できるなら,その人の好きなように扱って構いません.しかし,少しでも他人の目に触れる機会があるなら,一般化する方が好ましいと思います.
以下の例はあくまで例なので参考までに.
Bacillus subtilis の ftsz では,"bsu_ftsz"
Staphylococcus aureus の ftsz では "sau_ftsz" と名付けることにします.
今回はDNA配列とアミノ酸配列の両方とも必要なので,ファイル名の末尾に "NT" (DNA) や "AA" (アミノ酸) を付けると良いでしょう.
先述の通り,配列を保存する場合の拡張子は ".seq" をお勧めします.拡張子変更が難しければ,rtfでもtxtでも良いです.
もし日付を付けておきたければ,頭に "181128_" などと付けるのも良いかもしれません.
bsu_ftsz_nt.seq (bsu_ftszのDNA配列)
bsu_ftsz_aa.seq (bsu_ftszのアミノ酸配列)
sau_ftsz_nt.seq (sau_ftszのDNA配列)
sau_ftsz_aa.seq (sau_ftszのアミノ酸配列)
181128_sau_ftsz_aa.seq (日付ありの例)
4.まとめ
今回は配列情報の取得について解説しました.
自分は配列アライメントを行うときは必ずこのルートで行っているので,迷ってる方はこの流れに身を置いてみるのも一つと思います.
(この流れは実際に自分が学部生の頃に学生実験で習った方法です)
次回は,今回取得した配列を用いて配列アライメントを行います.
そのうえで配列アライメントとは何ぞや,と突っ込んで説明していこうかなと考えています.
今後ともどうぞよろしくお願いいたします.
この記事が気に入ったらサポートをしてみませんか?